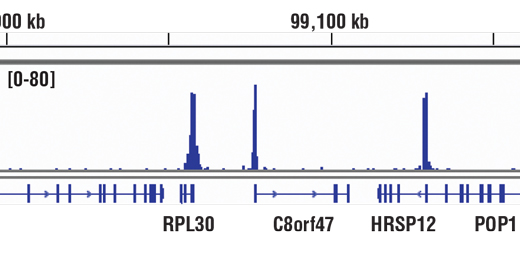
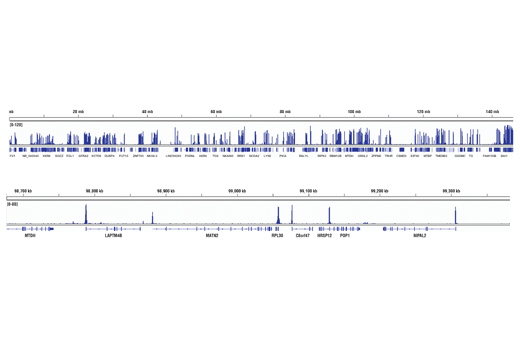
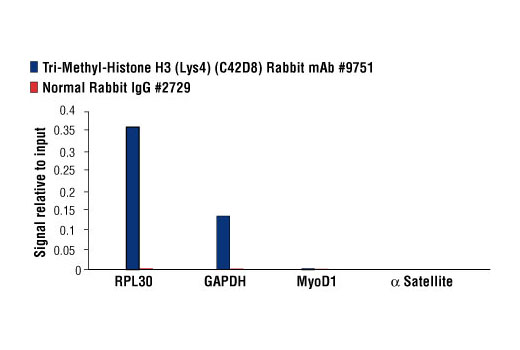
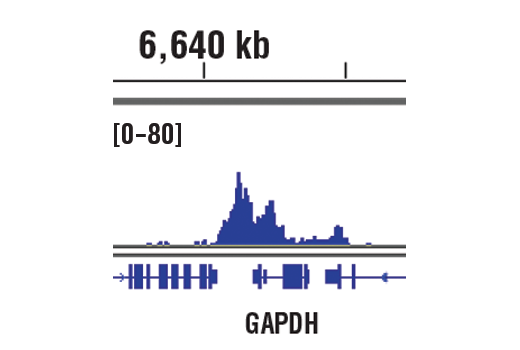
Recombinant antibodies offer several key advantages compared to traditional antibodies. These include superior lot-to-lot consistency, continuous supply, and animal-free manufacturing. As such, recombinant antibodies are seeing increased use for scientific research, especially as a means of addressing the ongoing reproducibility crisis.
Traditional polyclonal and monoclonal antibodies are the product of normal B cell development and genetic recombination. They are generated by immunizing an animal with an antigen to elicit an immune response. While polyclonal antibodies are secreted by many different B cell clones and recognize multiple antigenic epitopes, monoclonals originate from a single B cell clone and are specific for just one epitope.
Recombinant antibodies are monoclonal, but their production involves in vitro genetic manipulation. After cloning the antibody genes into an expression vector, this is then transfected into an appropriate host cell line for antibody expression. Mammalian cell lines are most commonly used for recombinant antibody production, although cell lines of bacterial, yeast, or insect origin are also suitable.
Because recombinant antibody production involves sequencing the antibody light and heavy chains, it is a highly controlled and reliable process. In contrast, hybridoma-based systems for producing monoclonal antibodies are subject to genetic drift and instability, increasing the potential for lot-to-lot variability or loss of antibody expression. Recombinant antibodies are highly consistent from lot to lot, thereby ensuring reproducible experimental results.
In vitro methods for producing antibodies are amenable to large-scale production, meaning antibody availability is unlikely to become a limiting factor. Moreover, since the recombinant antibody sequence is known, continuity of supply is assured; in situations where an antibody will be used to support large, long-term studies, this can be an especially critical factor.
Unlike traditional methods for antibody production, recombinant approaches avoid the need to use animals. Where polyclonal antibodies are purified directly from the serum of the immunized host, and monoclonals are purified from either hybridoma-derived tissue culture supernatant or ascites, recombinant antibodies are instead purified from the tissue culture supernatants of transfected host cell lines. Regardless of whether an antibody is polyclonal, monoclonal or recombinant, it must always be properly validated in the intended application prior to experimental use. At CST, we adhere to the Hallmarks of Antibody Validation™, six complementary strategies for determining the specificity, sensitivity, and functionality of an antibody in any given assay. By carefully tailoring these strategies to each antibody product, we guarantee that CST antibodies will work as expected, to help you achieve results you can trust.

| Cat. # | Size | Price | Inventory |
|---|---|---|---|
| 9751T | 20 µl | ||
| 9751S | 100 µl |
| REACTIVITY | H M R Mk Dm Sc |
| SENSITIVITY | Endogenous |
| MW (kDa) | 17 |
| Source/Isotype | Rabbit IgG |
Product Information
For optimal ChIP and ChIP-seq results, use 10 μl of antibody and 10 μg of chromatin (approximately 4 x 106 cells) per IP. This antibody has been validated using SimpleChIP® Enzymatic Chromatin IP Kits.
The CUT&RUN dilution was determined using CUT&RUN Assay Kit #86652.
The CUT&Tag dilution was determined using CUT&Tag Assay Kit #77552.
| Application | Dilution |
|---|---|
| Western Blotting | 1:1000 |
| Immunohistochemistry (Paraffin) | 1:1000 - 1:4000 |
| Immunofluorescence (Immunocytochemistry) | 1:200 - 1:800 |
| Flow Cytometry (Fixed/Permeabilized) | 1:400 - 1:1600 |
| Chromatin IP | 1:50 |
| Chromatin IP-seq | 1:50 |
| CUT&RUN | 1:50 |
| CUT&Tag | 1:50 |
Supplied in 10 mM sodium HEPES (pH 7.5), 150 mM NaCl, 100 µg/ml BSA, 50% glycerol and less than 0.02% sodium azide. Store at –20°C. Do not aliquot the antibody.
For a carrier free (BSA and azide free) version of this product see product #19776.
For western blots, incubate membrane with diluted primary antibody in 5% w/v BSA, 1X TBS, 0.1% Tween® 20 at 4°C with gentle shaking, overnight.
NOTE: Please refer to primary antibody product webpage for recommended antibody dilution.
From sample preparation to detection, the reagents you need for your Western Blot are now in one convenient kit: #12957 Western Blotting Application Solutions Kit
NOTE: Prepare solutions with reverse osmosis deionized (RODI) or equivalent grade water.
Load 20 µl onto SDS-PAGE gel (10 cm x 10 cm).
NOTE: Loading of prestained molecular weight markers (#59329, 10 µl/lane) to verify electrotransfer and biotinylated protein ladder (#7727, 10 µl/lane) to determine molecular weights are recommended.
NOTE: Volumes are for 10 cm x 10 cm (100 cm2) of membrane; for different sized membranes, adjust volumes accordingly.
* Avoid repeated exposure to skin.
posted June 2005
revised June 2020
Protocol Id: 10
NOTE: Prepare solutions with reverse osmosis deionized (RODI) or equivalent grade water.
NOTE: Do not allow slides to dry at any time during this procedure.
For Citrate: Heat slides in a microwave submersed in 1X citrate unmasking solution until boiling is initiated; follow with 10 min at a sub-boiling temperature (95°-98°C). Cool slides on bench top for 30 min.
|
RECOMMENDED DETECTION REAGENTS |
SignalStain® Boost IHC Detection Reagent (HRP, Rabbit) #8114 | SignalStain® Boost IHC Detection Reagent (AP, Rabbit) #18653 |
|---|---|---|
|
COMPATIBLE CHROMOGEN |
SignalStain® DAB Substrate Kit #8059 | SignalStain® Vibrant Red Alkaline Phosphatase Substrate Kit #76713 |
| SignalStain® Vivid Purple Peroxidase Substrate Kit #96632 | SignalStain® Ultra Blue Alkaline Phosphatase Substrate Kit #12824 | |
| SignalStain® Deep Black Peroxidase Substrate Kit #72986 | ||
| SignalStain® Radiant Yellow Peroxidase Substrate Kit #69644 |
NOTE: Use of detection reagents other than those specified in this protocol may require further optimization of the primary antibody to account for the different sensitivities of the detection reagents.
posted February 2010
revised June 2020
Protocol Id: 283
Achieve higher quality immunofluorescent images using the efficient and cost-effective, pre-made reagents in our #12727 Immunofluorescence Application Solutions Kit
NOTE: Prepare solutions with reverse osmosis deionized (RODI) or equivalent grade water.
Recommended Fluorochrome-conjugated Anti-Rabbit secondary antibodies:
NOTE: Cells should be grown, treated, fixed and stained directly in multi-well plates, chamber slides or on coverslips.
Aspirate liquid, then cover cells to a depth of 2–3 mm with 4% formaldehyde diluted in 1X PBS.
NOTE: Formaldehyde is toxic, use only in a fume hood.
NOTE: All subsequent incubations should be carried out at room temperature unless otherwise noted in a humid light-tight box or covered dish/plate to prevent drying and fluorochrome fading.
posted November 2006
revised November 2013
Protocol Id: 24
All reagents required for this protocol may be efficiently purchased together in our Intracellular Flow Cytometry Kit (Methanol) #13593, or individually using the catalog numbers listed below.
NOTE: Prepare solutions with reverse osmosis deionized (RODI) or equivalent grade water.
NOTE: When including fluorescent cellular dyes in your experiment (including viability dyes, DNA dyes, etc.), please refer to the dye product page for the recommended protocol. Visit www.cellsignal.com for a full listing of cellular dyes validated for use in flow cytometry.
NOTE: Adherent cells or tissue should be dissociated and in single-cell suspension prior to fixation.
NOTE: Optimal centrifugation conditions will vary depending upon cell type and reagent volume. Generally, 150-300g for 1-5 minutes will be sufficient to pellet the cells.
NOTE: If using whole blood, lyse red blood cells and wash by centrifugation prior to fixation.
NOTE: Antibodies targeting CD markers or other extracellular proteins may be added prior to fixation if the epitope is disrupted by formaldehyde and/or methanol. The antibodies will remain bound to the target of interest during the fixation and permeabilization process. However, note that some fluorophores (including PE and APC) are damaged by methanol and thus should not be added prior to permeabilization. Conduct a small-scale experiment if you are unsure.
NOTE: Count cells using a hemocytometer or alternative method.
posted July 2009
revised June 2020
实验步骤编号:404
特定产品: SimpleChIP® Plus Enzymatic Chromatin IP Kit (Magnetic Beads) #9005。
包括的试剂:
未包括的试剂:
| ! | 这就表示在实验流程中基于免疫沉淀制备物(IP 制备物)数量的容积改变是重要的一步。一份 IP 制备物是指 4 x 106 个组织培养细胞或 25 mg 离体的组织。 |
| !! | 这就表示,进行操作前稀释缓冲液是重要的一步。 |
| 安全停止 | 如果需要停止,这是实验步骤中的一个安全停止点。 |
当收获组织时,去除样品上不需要的材料,如脂肪和坏死组织。随后可以立即处理并交联组织,或在干冰上冷冻并储存于 -80°C 以待稍后处理。为获得最佳的染色质产率和 ChIP 结果,每次进行免疫沉淀法测试时,使用 25 mg 的组织。不同组织类型的染色质产率也不尽相同,并且一些组织在每次免疫沉淀法测试需要超过 25 mg。有关不同组织类型的预期染色质产率的更多信息,请参见附录 A。额外需要一份染色质样品进行染色质消化和浓度分析(第四部分)。如果需要,应处理额外五份染色质样品,以最优化染色质消化(附录 B)。
(!) 所有缓冲液的用量都应按照实验中 IP 制备物的数量成比例的增加。
为达到最佳染色质免疫沉淀法结果,每次免疫沉淀法使用大约 4 X 106 细胞进行实验(至少需要 12 X 106 个细胞以包含阳性和阴性对照)。对于 Hela 细胞,一次免疫沉淀相当于 15 cm 培养皿所含细胞(在 20 ml 生长培养基中的融合度为 90%)的一半。要进行染色质消化和浓度分析,应当处理一份额外样品(第四部分)。因为每种细胞类型都不相同,我们推荐您在实验中准备一盘额外的细胞,通过使用血球仪或细胞计数器确定细胞的数量。
(!) 所有缓冲液体积应根据使用的 15 cm 组织培养皿(或 20 ml 悬浮细胞)数量按比例增加。
(!) 所有缓冲液的用量都应按照实验中 IP 制备物的数量成比例的增加。
(!!) 重要提示:一旦溶解,将 1 M DTT 置于 -20℃ 下存放。
注意:为了获得最佳的 ChIP 结果,适宜大小和合适浓度的染色质非常关键。染色质过度消化可能会在 PCR 定量时削弱信号。染色质消化不充分可能导致背景信号增加和分辨率降低。向 IP 中添加染色质过少可能导致在 PCR 定量时信号削弱。优化染色质消化的实验步骤可以在附录 B 中找到。
为了获得最佳的 ChIP 结果,每次免疫沉淀使用大约 5 至 10 μg 消化的交联染色质(如第四部分中所测定)。这应当和来自 25 mg 分散的组织或 4 x 106 个组织培养细胞的单一 100μl IP 制备物大致相等。在添加抗体之前,通常将 100μl 消化的染色质稀释到 400μl 1X ChIP 缓冲液中。但是,如果每次 IP 需要 100 μl 以上的染色质,则交联的染色质制备物不需要按照下述方法进行稀释。可以直接向未稀释的染色质制备物中添加抗体以对染色质复合体进行免疫沉淀测试。
(!) 所有缓冲液的用量都应按照实验中免疫沉淀物的数量成比例的增加。
注意:Cell Signaling Technology 抗体在每份处于 1 和 2 ug 之间 IP 样品会取得最佳效果。当存在多份不同浓度的样品时,最好使阴性对照 Normal Rabbit IgG #2729 与最高抗体浓度相匹配。
(!) 所有缓冲液的用量都应按照实验中免疫沉淀物的数量成比例的增加。
| 引物长度: | 24 个核苷酸 |
| 最佳 Tm: | 60℃ |
| 最佳 GC: | 50% |
| 扩增子尺寸: | 150 至 200 bp(标准 PCR) |
| 80 至 160 bp(实时荧光定量 PCR) |
| 试剂 | 1 次 PCR 反应所需体积 (18 μl) |
|---|---|
| 无核酸酶的 H2O | 12.5 μl |
| 10X PCR 缓冲液 | 2.0 μl |
| 4mM dNTP 混合物 | 1.0 μl |
| 5 μM RPL30 引物 | 2.0 μl |
| Taq DNA 聚合酶 | 0.5 μl |
| a. | 初始变性 | 95℃ | 5 分钟 |
| b. | 变性 | 95℃ | 30 秒 |
| c. | 复性 | 62℃ | 30 秒 |
| d. | 延伸 | 72℃ | 30 秒 |
| e. | 重复步骤 b-d,共循环 34 次。 | ||
| f. | 终末延伸 | 72℃ | 5 分钟 |
| 试剂 | 1 次 PCR 反应所需体积 (18 μl) |
|---|---|
| 无核酸酶的 H2O | 6 μl |
| 5 μM RPL30 引物 | 2 μl |
| SimpleChIP® Universal qPCR Master Mix #88989 | 10 μl |
| a. | 初始变性 | 95℃ 3 分钟 |
| b. | 变性 | 95℃ 15 秒 |
| c. | 复性和延伸: | 60℃ 60 秒 |
| d. | 重复步骤 b 和 c,共循环 40 次。 | |
使用实时 PCR 仪的自带软件,分析定量 PCR 结果。或者,可以使用input样品百分数方法和以下所示的公式,手动计算 IP 效率。采用这种方法,从每次免疫沉淀获得的信号表述为占总input样品染色质的百分比。
输入百分比 = 2% x 2(C[T] 2% 输入样品 – C[T] IP 样品)
C[T] = CT = PCR 反应的阈周期
用这种试剂盒制备的免疫富集的 DNA 样品可用于 ChIP-seq 。要构建下游 NG 测序用 DNA 文库,请使用与您的下游测序平台相容的 DNA 文库制备实验步骤或试剂盒。对于 Illumina® 平台上测序,我们建议使用 DNA Library Prep Kit for Illumina® (ChIP-seq, CUT&RUN) #56795 及其相关索引引物 Multiplex Oligos for Illumina® (Single Index Primers) (ChIP-seq, CUT&RUN) #29580 或 Multiplex Oligos for Illumina® (Dual Index Primers) (ChIP-seq, CUT&RUN) #47538。
建议:
从组织样品收获交联的染色质时,组织类型之间的染色质产率可能显著不同。右表显示了用 25 mg 组织(类似于 4 x 106 个 Hela 细胞)制备的染色质预期产率和预期 DNA 浓度的范围,该产率和浓度按实验步骤第四部分的方法测定。对于每种组织类型,使用 Medimachine (BD Biosciences) 或 Dounce 匀浆器离散获得了相似数量的染色质。但是,与用通过 Dounce 匀浆器离散的组织制备和处理的染色质相比,用通过 Medimachine 离散的组织制备和处理的染色质通常具有更高的 IP 效率。强烈推荐 Dounce 匀浆器用于离散脑组织,原因是 Medimachine 无法将脑组织充分分离成单细胞悬液。为了获得最佳的 ChIP 结果,我们建议每次免疫沉淀使用 5 至 10 μg 消化的交联染色质,因此,对于某些组织,每次免疫沉淀可能需要收获超过 25 mg。
| 组织/细胞 | 染色质总产率 | 预期 DNA 浓度 |
|---|---|---|
| 脾 | 20-30 μg/25 mg 组织 | 200-300 μg/ml |
| 肝 | 10-15 μg/25 mg 组织 | 100-150 μg/ml |
| 肾 | 8-10 μg/25 mg 组织 | 80-100 μg/ml |
| 脑 | 2-5 μg/25 mg 组织 | 20-50 μg/ml |
| 心脏 | 2-5 μg/25 mg 组织 | 20-50 μg/ml |
| Hela | 10-15 μg/4 x 106 个细胞 | 100-150 μg/ml |
将交联的染色质 DNA 消化成 150-900 个碱基对长度的最佳条件高度取决于 Micrococcal Nuclease 对消化中所用组织量或细胞数目的比率。以下是确定特定组织或细胞类型的最佳消化条件的实验步骤。
| 问题 | 可能的原因 | 建议 |
|---|---|---|
| 1. 消化的染色质浓度过低。 | 添加至染色质消化的细胞数不足,或者胞核在消化后未完全裂解。 | 如果染色质制备物的 DNA 浓度接近于 50 μg/ml,则向每次 IP 中添加额外的染色质以确保每次 IP 至少产生 5 μg,然后继续实验。 在交联之前计算备用平板上细胞的数量,以确定准确的细胞数量,和/或在声波处理前后在显微镜下观察胞核,以证实胞核完全裂解。 |
| 2. 染色质消化不足且片段过大(大于 900 bp)。 | 细胞可能已经过度交联。交联超过 10 分钟可能会抑制染色质的消化。 向染色质消化中添加过多细胞或未添加足够的 Micrococcal Nuclease。 | 在甲醛浓度固定的情况下进行一段时间。将交联时间缩短至 10 分钟或更短。 在交联之前计算备用平板上细胞的数量,以确定准确的细胞数量;有关染色质消化的优化,参见附录 B。 |
| 3. 染色质过度消化并且片段过小(只有 150 bp 单核小体的长度)。在 PCR 定量期间,将染色质完全消化成单核小体长度的 DNA 可能削弱信号,尤其对于长度大于 150 bp 的扩增子更是如此。 | 添加至染色质消化的细胞不足或 Micrococcal Nuclease 过多。 | 在交联之前计算备用平板上细胞的数量,以确定准确的细胞数量;有关染色质消化的优化,参见附录 B。 |
| 4. 样品输入对照组 PCR 反应中无产物或产物很少。 | 添加至 PCR 反应的 DNA 不足或条件不是最佳。 PCR 扩增区域可能跨越无核小体的区域。 添加至 IP 反应的染色质不足或染色质过度消化。 | 向 PCR 反应中添加更多 DNA 或增加扩增循环次数。 使用来自交联并消化后的染色质的纯化 DNA,优化实验引物组的 PCR 条件。设计不同的引物组并将扩增子的长度减少到小于 150 碱基对(请参见第八部分中的引物设计建议)。 为获得最佳 ChIP 结果,每次 IP 添加 5-10 μg 染色质。参见上述问题 1 和 3 的建议。 |
| 5. 阳性对照组蛋白 H3-IP RPL30 PCR 反应中无产物。 | 添加至 IP 反应的染色质或抗体不足,或者 IP 孵育时间过短。 蛋白 G 微珠中染色质洗脱不完全。 | 确保向每次 IP 反应中添加 5-10 μg 染色质和 10 μl 抗体并和抗体一起孵育过夜。在添加蛋白 G 微珠后需额外孵育 2 小时。 在 65℃ 下将染色质从蛋白 G 微珠洗脱为最佳,同时频繁混合以保持微珠悬浮在溶液中。 |
| 6. 阴性对照 Rabbit IgG-IP 和阳性对照 Histone H3-IP PCR 反应中产物的数量相等。 | 添加至 IP 反应的染色质过多或不足。或者,添加至 IP 反应的抗体过多。 添加至 PCR 反应的 DNA 过多或扩增循环次数过多。 | 向每次 IP 反应中添加不超过 15 μg 的染色质和 10 μl Histone H3 Antibody。每次 IP 将 Normal Rabbit IgG 缩减至1 µl/IP。 向 PCR 反应中添加更少的 DNA 或减少 PCR 循环次数。在 PCR 的线性扩增阶段范围内分析 PCR 产物极为重要。否则,不能准确测量起始 DNA 数量的差异。 |
| 7. 实验抗体 IP PCR 反应中无产物。 | 添加至 PCR 反应的 DNA 不足。 添加至 IP 反应的抗体不足。 抗体不适用于 IP。 | 向 PCR 反应中添加更多 DNA 或增加扩增循环次数。 通常,需向 IP 反应中添加 1 至 5 µg 的抗体;但是,实际加入量很大程度上取决于各个抗体本身。 增加添加至 IP 反应的抗体量。寻找其他替代抗体。 |
发布时间 2011 年 12 月
修订时间 2022 年 4 月
实验步骤编号:82
特定产品: SimpleChIP® Plus Enzymatic Chromatin IP Kit (Magnetic Beads) #9005。
包括的试剂:
未包括的试剂:
当收获组织时,去除样品上不需要的材料,如脂肪和坏死组织。随后可以立即处理并交联组织,或在干冰上冷冻以待稍后处理。为获得最佳的染色质产率和 ChIP 结果,每次进行免疫沉淀法测试时,使用 25 mg 的组织。不同组织类型的染色质产率也不尽相同,并且一些组织在每次免疫沉淀法测试需要超过 25 mg。有关不同组织类型的预期染色质产率的更多信息,请参见附录 A。额外需要一份染色质样品进行染色质消化和浓度分析(第四部分)。
为了获得最佳 ChIP 结果,对于每次将要进行免疫沉淀法测试,使用大约 4 X 106 个细胞。对于 Hela 细胞,这相当于 15 cm 培养皿所含细胞(在 20 ml 生长培养基中的融合度为 90 %)的一半。要进行染色质消化和浓度分析,应当处理一份额外样品(第四部分)。在实验中额外使用一培养皿的细胞,以便使用血细胞计数器测量细胞数目。
一份免疫沉淀制备物(IP 制备物)是指 25 mg 离散的组织或 4 x 106 个组织培养细胞。
配制 1 M DTT (192.8 mg DTT #7016 + 1.12 ml dH2O)。确保 DTT 晶体完全溶解。
重要提示:一旦溶解,将 1 M DTT 置于 -20℃ 下存放。
注意:为了获得最佳的 ChIP 结果,适宜大小和合适浓度的染色质非常关键。染色质过度消化可能会在 PCR 定量时削弱信号。染色质消化不充分可能导致背景信号增加和分辨率降低。向 IP 中添加染色质过少可能导致在 PCR 定量时信号削弱。优化染色质消化的实验步骤可以在附录 B 中找到。
为了获得最佳的 ChIP 结果,每次免疫沉淀使用大约 5 至 10 μg 消化的交联染色质(如第四部分中所测定)。这应当和来自 25 mg 分散的组织或 4 x 106 个组织培养细胞的单一 100μl IP 制备物大致相等。在添加抗体之前,通常将 100μl 消化的染色质稀释到 400μl 1X ChIP 缓冲液中。但是,如果每次 IP 需要 100 μl 以上的染色质,则交联的染色质制备物不需要按照下述方法进行稀释。可以直接向未稀释的染色质制备物中添加抗体以对染色质复合体进行免疫沉淀测试。
注意:Cell Signaling Technology 抗体在每份处于 1 和 2 ug 之间 IP 样品会取得最佳效果。当存在多份不同浓度的样品时,最好使阴性对照 Normal Rabbit IgG #2729 与最高抗体浓度相匹配。
| 引物长度: | 24 个核苷酸 |
| 最佳 Tm: | 60℃ |
| 最佳 GC: | 50% |
| 扩增子尺寸: | 150 至 200 bp(标准 PCR) |
| 80 至 160 bp(实时荧光定量 PCR) |
| 试剂 | 1 次 PCR 反应所需体积 (18 μl) |
|---|---|
| 无核酸酶的 H2O | 12.5 μl |
| 10X PCR 缓冲液 | 2.0 μl |
| 4mM dNTP 混合物 | 1.0 μl |
| 5 μM RPL30 引物 | 2.0 μl |
| Taq DNA 聚合酶 | 0.5 μl |
| a. | 初始变性 | 95℃ | 5 分钟 |
| b. | 变性 | 95℃ | 30 秒 |
| c. | 复性 | 62℃ | 30 秒 |
| d. | 延伸 | 72℃ | 30 秒 |
| e. | 重复步骤 b-d,共循环 34 次。 | ||
| f. | 终末延伸 | 72℃ | 5 分钟 |
| 试剂 | 1 次 PCR 反应所需体积 (18 μl) |
|---|---|
| 无核酸酶的 H2O | 6 μl |
| 5 μM RPL30 引物 | 2 μl |
| SYBR-Green Reaction Mix | 10 μl |
| a. | 初始变性 | 95℃ 3 分钟 |
| b. | 变性 | 95℃ 15 秒 |
| c. | 复性和延伸: | 60℃ 60 秒 |
| d. | 重复步骤 b 和 c,共循环 40 次。 | |
使用实时 PCR 仪的自带软件,分析定量 PCR 结果。或者,可以使用input样品百分数方法和以下所示的公式,手动计算 IP 效率。采用这种方法,从每次免疫沉淀获得的信号表述为占总input样品染色质的百分比。
输入百分比 = 2% x 2(C[T] 2% 输入样品 – C[T] IP 样品)
C[T] = CT = PCR 反应的阈周期
用这种试剂盒制备的免疫富集的 DNA 样品可用于 ChIP-seq 。要构建下游 NG 测序用 DNA 文库,请使用与您的下游测序平台相容的 DNA 文库制备实验步骤或试剂盒。对于 Illumina® 平台上测序,我们建议使用 DNA Library Prep Kit for Illumina® (ChIP-seq, CUT&RUN) #56795 及其相关索引引物 Multiplex Oligos for Illumina® (Single Index Primers) (ChIP-seq, CUT&RUN) #29580 或 Multiplex Oligos for Illumina® (Dual Index Primers) (ChIP-seq, CUT&RUN) #47538。
建议:
从组织样品收获交联的染色质时,组织类型之间的染色质产率可能显著不同。右表显示了用 25 mg 组织(类似于 4 x 106 个 Hela 细胞)制备的染色质预期产率和预期 DNA 浓度的范围,该产率和浓度按实验步骤第四部分的方法测定。对于每种组织类型,使用 Medimachine (BD Biosciences) 或 Dounce 匀浆器离散获得了相似数量的染色质。但是,与用通过 Dounce 匀浆器离散的组织制备和处理的染色质相比,用通过 Medimachine 离散的组织制备和处理的染色质通常具有更高的 IP 效率。强烈推荐 Dounce 匀浆器用于离散脑组织,原因是 Medimachine 无法将脑组织充分分离成单细胞悬液。为了获得最佳的 ChIP 结果,我们建议每次免疫沉淀使用 5 至 10 μg 消化的交联染色质,因此,对于某些组织,每次免疫沉淀可能需要收获超过 25 mg。
| 组织/细胞 | 染色质总产率 | 预期 DNA 浓度 |
|---|---|---|
| 脾 | 20-30 μg/25 mg 组织 | 200-300 μg/ml |
| 肝 | 10-15 μg/25 mg 组织 | 100-150 μg/ml |
| 肾 | 8-10 μg/25 mg 组织 | 80-100 μg/ml |
| 脑 | 2-5 μg/25 mg 组织 | 20-50 μg/ml |
| 心脏 | 2-5 μg/25 mg 组织 | 20-50 μg/ml |
| Hela | 10-15 μg/4 x 106 个细胞 | 100-150 μg/ml |
将交联的染色质 DNA 消化成 150-900 个碱基对长度的最佳条件高度取决于 Micrococcal Nuclease 对消化中所用组织量或细胞数目的比率。以下是确定特定组织或细胞类型的最佳消化条件的实验步骤。
| 问题 | 可能的原因 | 建议 |
|---|---|---|
| 1. 消化的染色质浓度过低。 | 添加至染色质消化的细胞数不足,或者胞核在消化后未完全裂解。 | 如果染色质制备物的 DNA 浓度接近于 50 μg/ml,则向每次 IP 中添加额外的染色质以确保每次 IP 至少产生 5 μg,然后继续实验。 在交联之前计算备用平板上细胞的数量,以确定准确的细胞数量,和/或在声波处理前后在显微镜下观察胞核,以证实胞核完全裂解。 |
| 2. 染色质消化不足且片段过大(大于 900 bp)。 | 细胞可能已经过度交联。交联超过 10 分钟可能会抑制染色质的消化。 向染色质消化中添加过多细胞或未添加足够的 Micrococcal Nuclease。 | 在甲醛浓度固定的情况下进行一段时间。将交联时间缩短至 10 分钟或更短。 在交联之前计算备用平板上细胞的数量,以确定准确的细胞数量;有关染色质消化的优化,参见附录 B。 |
| 3. 染色质过度消化并且片段过小(只有 150 bp 单核小体的长度)。在 PCR 定量期间,将染色质完全消化成单核小体长度的 DNA 可能削弱信号,尤其对于长度大于 150 bp 的扩增子更是如此。 | 添加至染色质消化的细胞不足或 Micrococcal Nuclease 过多。 | 在交联之前计算备用平板上细胞的数量,以确定准确的细胞数量;有关染色质消化的优化,参见附录 B。 |
| 4. 样品输入对照组 PCR 反应中无产物或产物很少。 | 添加至 PCR 反应的 DNA 不足或条件不是最佳。 PCR 扩增区域可能跨越无核小体的区域。 添加至 IP 反应的染色质不足或染色质过度消化。 | 向 PCR 反应中添加更多 DNA 或增加扩增循环次数。 使用来自交联并消化后的染色质的纯化 DNA,优化实验引物组的 PCR 条件。设计不同的引物组并将扩增子的长度减少到小于 150 碱基对(请参见第八部分中的引物设计建议)。 为获得最佳 ChIP 结果,每次 IP 添加 5-10 μg 染色质。参见上述问题 1 和 3 的建议。 |
| 5. 阳性对照组蛋白 H3-IP RPL30 PCR 反应中无产物。 | 添加至 IP 反应的染色质或抗体不足,或者 IP 孵育时间过短。 蛋白 G 微珠中染色质洗脱不完全。 | 确保向每次 IP 反应中添加 5-10 μg 染色质和 10 μl 抗体并和抗体一起孵育过夜。在添加蛋白 G 微珠后需额外孵育 2 小时。 在 65℃ 下将染色质从蛋白 G 微珠洗脱为最佳,同时频繁混合以保持微珠悬浮在溶液中。 |
| 6. 阴性对照 Rabbit IgG-IP 和阳性对照 Histone H3-IP PCR 反应中产物的数量相等。 | 添加至 IP 反应的染色质过多或不足。或者,添加至 IP 反应的抗体过多。 添加至 PCR 反应的 DNA 过多或扩增循环次数过多。 | 向每次 IP 反应中添加不超过 15 μg 的染色质和 10 μl Histone H3 Antibody。每次 IP 将 Normal Rabbit IgG 缩减至1 µl/IP。 向 PCR 反应中添加更少的 DNA 或减少 PCR 循环次数。在 PCR 的线性扩增阶段范围内分析 PCR 产物极为重要。否则,不能准确测量起始 DNA 数量的差异。 |
| 7. 实验抗体 IP PCR 反应中无产物。 | 添加至 PCR 反应的 DNA 不足。 添加至 IP 反应的抗体不足。 抗体不适用于 IP。 | 向 PCR 反应中添加更多 DNA 或增加扩增循环次数。 通常,需向 IP 反应中添加 1 至 5 µg 的抗体;但是,实际加入量很大程度上取决于各个抗体本身。 增加添加至 IP 反应的抗体量。寻找其他替代抗体。 |
发布时间 2011 年 12 月
修订时间 2022 年 4 月
实验步骤编号:1184
| ! | ! 表示实验步骤中需要根据进行的 CUT&RUN 反应次数来调整体积的重要步骤。 |
| !! | !! 表示需要在操作前稀释缓冲液的一个重要步骤。 |
| 安全停止 | 如果需要停止,这是实验步骤中的一个安全停止点。 |
对于大多数细胞类型,活细胞可用于 CUT&RUN 检测,以产生强大的组蛋白、转录因子和辅因子富集。就某些对伴刀豆球蛋白 A 脆弱或敏感的细胞类型,略微固定细胞有助于保存细胞并保持它们完整。此外,如果使用新鲜细胞未观察到明显信号,固定可能有助于促进低丰度和/或弱结合转录因子和辅因子的富集。请注意,细胞的过度固定会抑制 CUT&RUN 检测。
我们的 CUT&RUN 分析适用于广泛的细胞或组织。如实验步骤中定义,一个 CUT&RUN 反应可包含 5,000 至 个 250,000 细胞或 1 至 5 mg 的组织。整个实验步骤中使用的缓冲液体积无需根据每次反应的细胞或组织数量进行调整,只要在此范围内即可。当需要时,缓冲液体积确实需要根据正在执行的反应数量按比例增加。如果可能,我们建议每次反应使用 100,000 个细胞或 1 mg 组织。如果细胞数量有限,我们建议每个反应至少使用 5,000 到 10,000 个细胞进行组蛋白修饰,每个反应至少使用 10,000 到 20,000 个细胞进行转录因子或辅因子。
注意: 推荐用于细胞透化的毛地黄皂苷的量是过量的,应该足以用于大多数细胞系和组织类型的通透。然而,并非所有细胞系和组织对毛地黄皂苷都表现出相同的敏感性。如果您的特定细胞系或组织在推荐的毛地黄皂苷浓度下不起作用,您可以按照附录 A 中提供的实验步骤来优化条件。毛地黄皂苷处理应导致 >90% 的细胞群通透。
! 所有缓冲液的体积都应根据正在进行的 CUT&RUN 反应次数按比例增加。
注意: 应在室温下连续进行活细胞(无固定)制备步骤,以尽量减少对细胞的压力。为了尽量减少 DNA 碎片,在重悬过程中避免剧烈涡旋和样品起泡。为 CUT&RUN 制备活细胞时,我们建议在制备细胞之前准备伴刀豆球蛋白 A 珠子(第 II 部分,步骤 1 到 5),以尽量减少细胞在珠子制备过程中停留的时间。活化的珠子可以储存在冰上直到准备使用。
注:对于贴壁细胞,首先需要使用胰蛋白酶将细胞从培养皿中分离出来,并用至少 3 体积的组织培养基中和。我们不建议从培养皿中刮取细胞,因为这会压迫甚至裂解细胞。应使用血细胞计数器或其他细胞计数器对细胞进行计数,以确保用于实验的细胞数量正确。
注:处理低细胞数(<100,000 总细胞)的挑战在于,离心后的细胞颗粒并不总是肉眼可见,因此很容易在洗涤步骤中丢失细胞。因此,当处理低细胞数时,我们建议跳过下面的洗涤步骤 3 到 5。伴刀豆球蛋白 A 珠与细胞的结合耐受结合反应中有 40% 的细胞培养基。因此,在步骤 2 中对细胞悬浮液进行初始离心后,可以去除大部分上清液,每次反应留下 ≤40 µl 细胞培养基。然后在步骤 6 中,向细胞悬液中加入足够的 1X 洗涤缓冲液(+ 亚精胺 + PIC),使每次反应的总体积达到 100 µl。
注意:在随后的实验步骤中,将在 55°C 下孵育 input 样品,因此建议使用一根可封盖的 1.5 ml 试管,以减少孵育过程出现蒸发。
注:固定细胞制备需要以下试剂,不包含在此试剂盒中:37% 甲醛或 16% 无甲醇的甲醛 #12606,甘氨酸溶液 (10X) #7005 和 10% SDS 溶液 #20533。
! 所有缓冲液的体积都应根据正在进行的 CUT&RUN 反应次数按比例增加。
注释:对于贴壁细胞系,首先需要使用胰蛋白酶将细胞从培养皿中分离出来,并用至少 3 体积的培养基中和。我们不建议从培养皿中刮取细胞,因为这会压迫甚至裂解细胞。应使用血细胞计数器或其他细胞计数器对细胞进行计数,以确保用于实验的细胞数量正确。
注:处理低细胞数(<100,000 总细胞)的挑战在于,离心后的细胞颗粒并不总是肉眼可见,因此很容易在洗涤步骤中丢失细胞。在这种情况下,我们不建议冷冻细胞沉淀物。此外,当处理这些低细胞数时,我们建议跳过下面的洗涤步骤 5 到 7。伴刀豆球蛋白 A 珠与细胞的结合耐受结合反应中有 40% 的细胞培养基。因此,在步骤 4 中对细胞悬浮液进行初始离心后,可以去除大部分上清液,每次反应留下 ≤40 µl 细胞培养基。然后在步骤 8 中,向细胞悬液中加入足够的 1X 洗涤缓冲液(+ 亚精胺 + PIC),使每次反应的总体积达到 100 µl。
注意:在随后的实验步骤中,将在 55°C 下孵育 input 样品,因此建议使用一根可封盖的 1.5 ml 试管,以减少孵育过程出现蒸发。
对于大多数组织类型,1 mg 轻微固定的组织(0.1% 甲醛,2 分钟)可以产生强大的组蛋白、转录因子和辅因子富集。富集组蛋白修饰不需要甲醛固定。然而,许多转录因子和辅因子确实需要对组织进行光固定以获得最佳结果。一些低丰度和/或弱结合转录因子和辅助因子可能需要中等固定(0.1% 甲醛,10 分钟)以获得最佳结果。此外,在使用具有挑战的组织类型(如纤维组织)时,中等固定可能会改善结果。请注意,过度固定会抑制 CUT&RUN 检测。固定的组织样本在使用前可以在 -80°C 下冷冻长达 6 个月。
注:为 CUT&RUN 准备新鲜组织(无固定)时,我们建议在制备组织之前制备伴刀豆球蛋白 A 珠(第二部分,步骤 1 到 5),以尽量减少细胞在珠子制备过程中停留的时间。活化的珠子可以储存在冰上直到准备使用。
注:固定组织制备需要以下试剂,不包含在此试剂盒中:37% 甲醛或 16% 无甲醇的甲醛 #12606、磷酸盐缓冲盐水 (PBS) #9872、甘氨酸溶液 (10X) #7005 和 10% SDS 溶液 #20533。
! 所有缓冲液的体积都应根据正在进行的 CUT&RUN 反应次数按比例增加。
注意:对于某些转录因子或辅因子,或对于诸如纤维组织之类的困难组织类型,每个反应最多可使用 5 mg 组织而无需按比例放大试剂。
注意:我们建议对组织进行稍稍固定,因为这种条件最适合大多数组织类型和蛋白质靶标。但是,如果需要新鲜组织,请跳过步骤 3 到 8 并立即进行步骤 9。
注意:此体积的固定溶液足以容纳多达 50 mg 的组织。如果处理 >50 mg,则在步骤 7 中相应地增加固定溶液和 1X PBS + PIC 溶液。
注意:对于困难的组织类型(如纤维组织)或低丰度和/或弱结合转录因子或辅因子,将甲醛固定延长至 10 分钟可能会改善结果。
注意:在随后的实验步骤中,将在 55°C 下孵育 input 样品,因此建议使用一根可封盖的 1.5 ml 试管,以减少孵育过程出现蒸发。
! 所有缓冲液的体积都应根据正在进行的 CUT&RUN 反应次数按比例增加。
NOTE: Digitonin Solution #16359 should be stored at -20°C. 请在使用过程中将其保存在冰上,并在实验完成后保存在 -20°C 下。
注意:避免将伴刀豆球蛋白 A 珠悬浮液涡旋,因为反复涡旋可能会使伴刀豆球蛋白 A 从珠子中移位。
注意:为避免珠子丢失,请使用移液管去除液体。请勿使用真空设备抽吸。
注意:如果在细胞或组织制备之前制备伴刀豆球蛋白 A 珠子(如建议用于活细胞和新鲜组织),则活化的珠子可以储存在冰上直至使用。
注意:伴刀豆球蛋白 A 珠子可能会结块或粘附在试管壁上。上下吹打来重悬珠子。无需摇动或振摇样品样管。
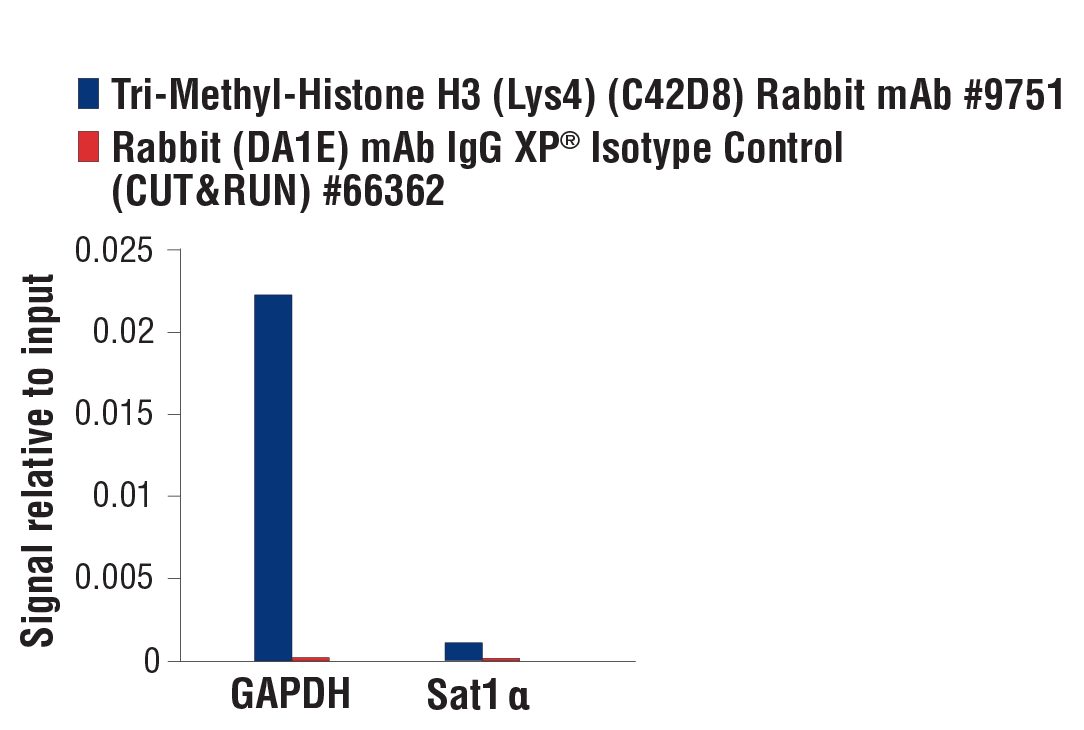
注意:CUT&RUN 需要的抗体量不定,应由使用者确定。对于阳性对照 Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit mAb #9751,向样品中添加 2 µl 抗体。对于阴性对照 Rabbit (DA1E) mAb IgG XP® Isotype Control (CUT&RUN) #66362,向样品中添加 5 µl。我们强烈建议使用阴性对照抗体,而不是非抗体对照,因为后者会导致高水平的非特异性 MNase 消化和高背景信号。我们建议尽可能使用 input 样本与 qPCR 和 NG-seq 分析进行比较。
注意:伴刀豆球蛋白 A 珠子可能会结块或粘附在试管壁上。上下吹打来重悬珠子。无需摇动或振摇样品样管。
! 所有缓冲液的体积都应根据正在进行的 CUT&RUN 反应次数按比例增加。
注意:洋地黄皂苷溶液 #16359 应保存在 -20°C 下。请在使用过程中将其保存在冰上,并在实验完成后保存在 -20°C 下。
NOTE: The Digitonin Buffer prepared here will be used for both Section III and IV.
注意:Concanavalin A 磁珠可能会结块或粘附在试管壁上。上下吹打来重悬珠子。无需摇动或振摇样品样管。
! 所有缓冲液的体积都应根据正在进行的 CUT&RUN 反应次数按比例增加。
注意:洋地黄皂苷溶液 #16359 应保存在 -20°C 下。请在使用过程中将其保存在冰上,并在实验完成后保存在 -20°C 下。
可选:如需对样品进行标准校对,则可将 Sample Normalization Spike-In DNA 添加到 1X Stop Buffer 中(例如,见第七部分的图 8)。对于 qPCR 分析,我们建议每次反应加入 5 µl (5 ng) Spike-In DNA。对于 NG-seq 分析,我们建议用 Nuclease-free Water #12931 稀释 Sample Normalization Spike-In DNA 100 倍,每次反应加入 5 µl (50 pg) Spike-In DNA。当每次反应使用 100,000 个细胞或 1 mg 组织时,这可确保标准化读数约为总测序读数的 0.5%。如果每个反应使用多于或少于 100,000 个的细胞或 1 mg 组织,按比例放大或缩小样本标准化 Spike-In DNA 的体积,以将标准化读数调整到总读数的 0.5% 左右。
注意:应在 4°C 的冷却块或冰箱中进行消化。冰的温度可低至 0°C,这可能会限制消化并降低信号。无需摇动或振摇样品样管。
注意:如果使用活细胞或新鲜组织(未固定)进行 CUT&RUN 测定,跳过步骤 14-15,然后立即进行步骤 16。
注意:在随后的实验步骤中,将在 65°C 下孵育 input 样品,因此建议使用一根可封盖的 2 ml 试管,以减少孵育过程出现蒸发。
注意:如果样品没有预热到室温,SDS 可能会从溶液中沉淀出来。
输入 DNA 的片段化是与下游 NG 测序兼容所必需的,但对于下游 qPCR 分析则不是必需的。如果没有超声波仪,我们建议使用未片段化的输入 DNA 进行 qPCR 分析;然而,输入 DNA 应使用苯酚/氯仿提取和乙醇沉淀进行纯化,因为未片段化输入 DNA 的尺寸太大而无法使用 DNA 离心柱进行纯化。如果没有超声波仪并且需要进行下游 NG 测序分析,则可以使用 CUT&RUN 正常 IgG 抗体样品作为阴性对照,尽管这并不理想,因为正常的富含 IgG 的样品可能会显示非特异性 DNA 富集。或者,使用 MNase 的输入 DNA 片段化实验步骤可在 https://cst-science.com/CUT-RUN-input-digestion 获得。
! 所有缓冲液的体积都应根据正在制备的input样品的数量按比例增加。
注意:超声处理条件可能需要根据 附录 B 中的步骤通过测试不同的超声仪功率设置和/或超声处理持续时间来凭经验确定。最佳超声处理条件将产生大小从 100-600 bp 不等的染色质片段。使用设置为 6 且配有 1/8 英寸探针的 VirTis Virsonic 100 超声波均质器/超声波仪以 5 组 15 秒脉冲进行超声处理,可充分碎裂input染色质。
如 VI - A 部分所述,使用 DNA 离心柱可从input和富集染色质样品中纯化 DNA,或如第 VI - B 部分所述,使用苯酚/氯仿提取后再行乙醇沉淀方法进行纯化。使用 DNA 离心柱纯化简单快速,可很好地回收 35 bp 以上的 DNA 片段(图 7A,泳道 2)。苯酚/氯仿提取之后再行乙醇沉淀更加困难,但能很好地回收 35 bp 以下的 DNA 片段(图 7A,泳道 3);但如图 7B 所示,大多数在 CUT&RUN 检测中产生的 DNA 片段大于 35 bp。因此,DNA 离心柱提供一种快速简单的方法来纯化 > 98% 的所有 CUT&RUN DNA 片段。
在 NG-seq 分析之前,使用基于 picogreen 的 DNA 定量测定法可以对纯化的 DNA 进行定量。对于含 100,000 个细胞的 CUT&RUN 反应,一次转录因子和辅因子的 CUT&RUN 反应中,预期 DNA 产量为 0.5-10 ng,而在一次组蛋白修饰反应中则为 1-20 ng。
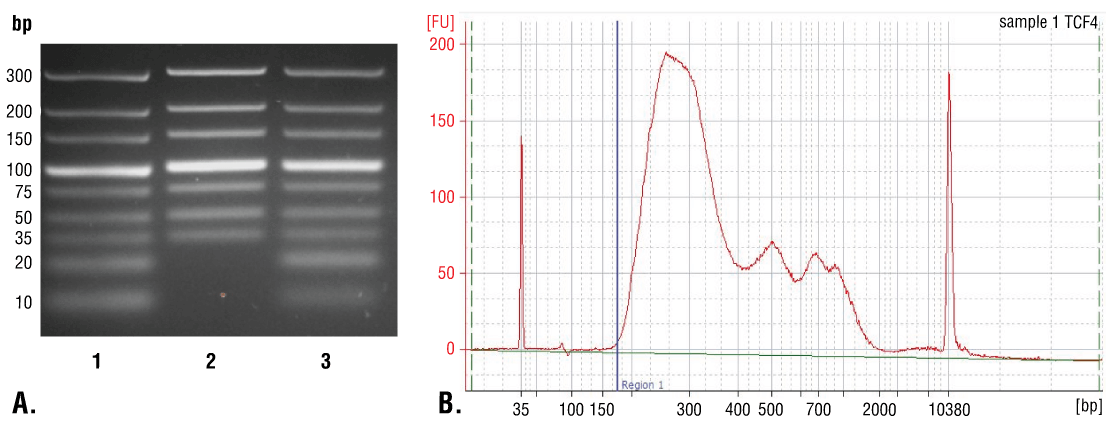
图 7. 比较使用离心柱或苯酚/氯仿提取后再行乙醇沉淀方法进行的 DNA 纯化。(A) 将低量程 DNA 标准品混合物(泳道 1,未纯化)使用 DNA Purification Buffers and Spin Columns (ChIP, CUT&RUN,CUT&Tag) #14209(泳道 2)或苯酚/氯仿提取后接乙醇沉淀法(泳道 3)纯化,并通过在 4% 琼脂糖凝胶上电泳来分离。如图所示,苯酚/氯仿提取后再行乙醇沉淀能有效回收所有大小的 DNA 片段,而 DNA 离心柱仅能回收 ≥ 35 bp 的 DNA 片段。(B) 在使用 TCF4/TCF7L2 (C48H11) Rabbit mAb #2569 的 CUT&RUN 检测中使用苯酚/氯仿提取后再行乙醇沉淀方法来纯化 DNA。使用一台Bioanalyzer (Agilent Technologies) 分析文库中 DNA 片段的大小。在构建期间添加到文库中的接头蛋白和条码序列的片段长度为 140 bp。因此,文库制备后,起始 35 bp 的 DNA 片段长度将变为 175 bp(图中用蓝色垂直线标注)。如图所示,总 CUT&RUN 富集 DNA 片段中不到 2% 短于 175 bp(起始长度 35 bp),提示 DNA 纯化离心柱足以捕获 > 98% 的总 CUT&RUN DNA 片段。
NOTE: DNA can be purified from input and enriched chromatin samples using the DNA Purification Buffers and Spin Columns (ChIP, CUT&RUN, CUT&Tag) #14209 (not included in this kit) and the modified protocol below. 步骤 1-5 已修改,以符合向 300 µl input样品和富集染色质样品中添加 5 倍体积 (1.5 ml) DNA 结合缓冲液的要求。
注意:1 体积样品应使用 5 体积的 DNA 结合缓冲液。
注意:以下试剂是苯酚/氯仿提取和乙醇沉淀所必需的,不包含在本试剂盒中:苯酚/氯仿/异戊醇 (25:24:1)、氯仿/异戊醇 (24:1)、3M 乙酸钠 (pH 5.2)、20 mg/ml 糖原、100% 乙醇、70% 乙醇和 1X TE 缓冲液或无核酸酶水 #12931。
注意:如果进行样品标准化,仅CUT&RUN 样品使用Sample Normalization Primer Set分析 。input DNA 不包含Normalization Spike-In DNA。
| 试剂 | 1 次 PCR 反应所需体积 (18 μl) |
|---|---|
| Nuclease-free H2O #12931 | 6 μl |
| 5 µM 引物 | 2 μl |
| SimpleChIP® Universal qPCR Master Mix #88989 | 10 μl |
| a. | 初始变性 | 95°C,3 分钟 |
| b. | 变性 | 95°C,15 秒钟 |
| c. | 退火和延伸 | 60°C,60 秒钟 |
| d. | 重复步骤 b 和 c,共循环 40 次。 |
| 样品标准化引物组的 C[T] 值 | **qPCR 的标准化系数 | 标准化之前的信号(步骤 5 中计算出的输入百分比) | 标准化之后的信号 | |
| 样品 1 | 23.31 | 2(23.31-23.31)=1.00 | 24.4% | 24.4%/1.00=24.4% |
| 样品 2 | 24.24 | 2(23.31-24.24)=0.52 | 12.0% | 12.0%/0.52=23.1% |
| 样品 3 | 25.08 | 2(23.31-25.08)=0.29 | 6.28% | 6.28%/0.29=21.7% |
| 样品 4 | 26.30 | 2(23.31-26.30)=0.13 | 2.72% | 2.72%/0.13=20.9% |
**qPCR 的标准化系数 = 2(C[T] 选定样品 – C[T] 其他样品)
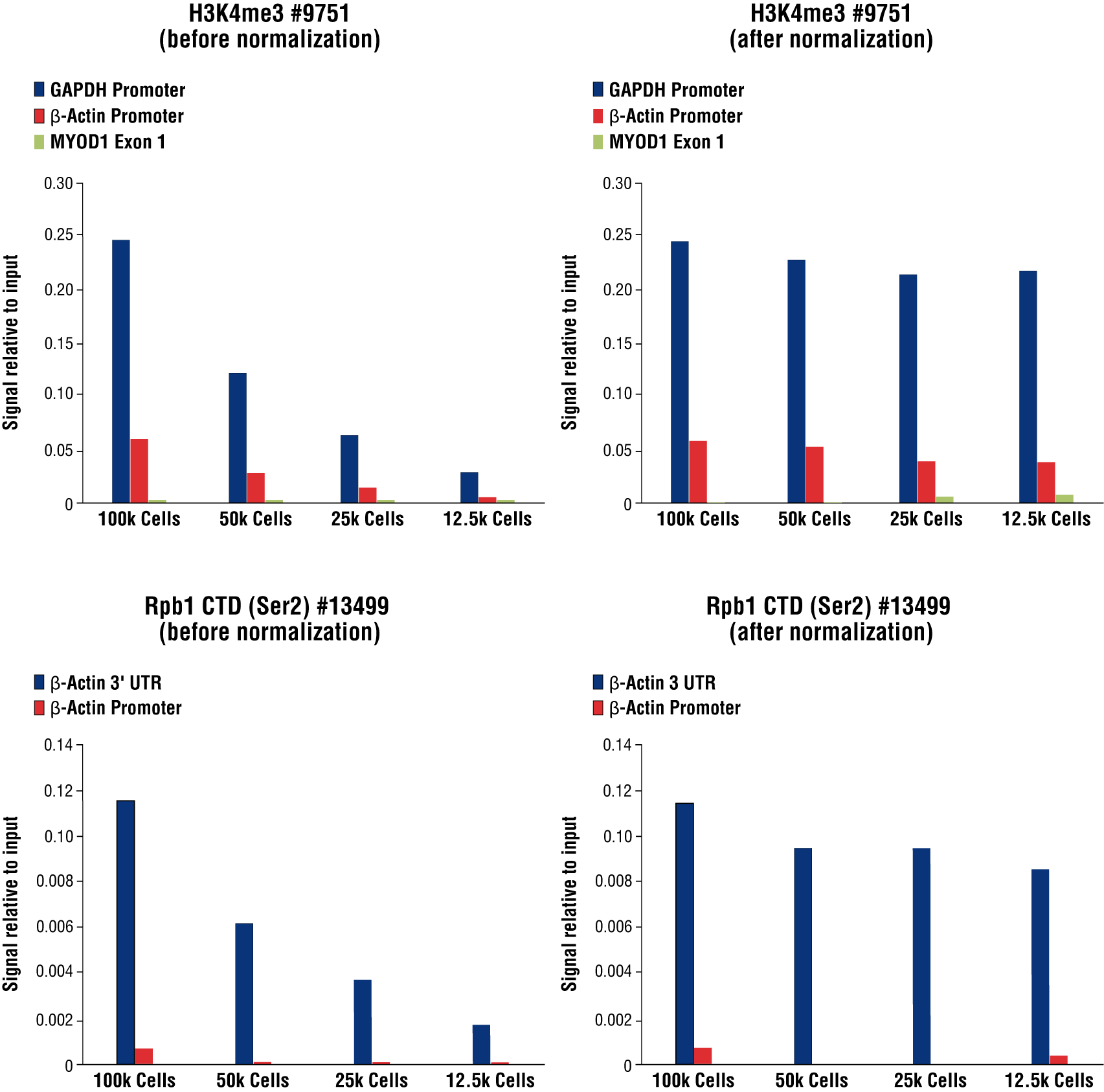
图 8. 应用添加的Spike-In DNA对 CUT&RUN 信号进行 qPCR 标准化分析。对数量渐减的 HCT116 细胞和 Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit mAb #9751 (上小图)或 Phospho-Rpb1 CTD (Ser2) (E1Z3G) Rabbit mAb #13499(下小图)进行 CUT&RUN 检测。使用 SimpleChIP® Human GAPDH Exon 1 Primers #5516、SimpleChIP® Human β-Actin Promoter Primers #13653、SimpleChIP® Human Β-Actin 3' UTR Primers #13669 和 SimpleChIP® Human MyoD1 Exon 1 Primers #4490 进行实时 PCR 来对富集的 DNA 进行定量。 每份样品中免疫沉淀 DNA 的数量表示为与 100,000 个细胞的input染色质总量相对应的信号。左图中为非标准化的富集结果。在每次反应中,按照与起始细胞数量成比例的方式添加 Sample Normalization Spike-In DNA。根据每份样品中添加 DNA 所产生的 qPCR 信号差异,将 CUT&RUN 信号标准化为含 100,000 个细胞的样品。右图显示了标准化后的富集。
用这种试剂盒制备的免疫富集 DNA 样品直接兼容 NG-seq。要构建下游 NG-seq DNA 文库,请使用与您的下游测序平台兼容的 DNA 文库制备实验步骤或试剂盒。对于在 Illumina® 平台上测序,我们建议按照 CUT&RUN DNA 的实验步骤,使用 DNA Library Prep Kit for Illumina® (ChIP-seq, CUT&RUN) #56795 和 Multiplex Oligos for Illumina® (ChIP-seq, CUT&RUN) #29580 或 #47538 进行测序。
| 与酵母对应的unique reads数 | NGS 的标准化系数 | 标准化之前与测试参考基因组对应的unique reads数 | NGS 的标准化系数 = 选定样品的酵母 reads数/其他样品的酵母unique reads数 | |
| 样品 1 | 219,275 | 219,275/219,275 = 1.00 | 5,077,747 | 5,077,747 X 1.00 = 5,077,747 |
| 样品 2 | 411,915 | 219,275/411,915 = 0.53 | 9,896,671 | 9,896,671 X 0.53 = 5,268,306 |
| 样品 3 | 816,235 | 219,275/816,235 = 0.27 | 17,842,773 | 17,842,773 X 0.27 = 4,793,320 |
| 样品 4 | 1,120,826 | 219,275/1,120,826 = 0.20 | 23,836,679 | 23,836,679 X 0.20 = 4,663,339 |
NGS 的标准化系数 = 选定样品的酵母unique reads数/其他样品的酵母unique reads数
在 CUT&RUN 实验步骤中,向缓冲液中添加洋地黄皂苷能促进细胞膜透化以及一抗和 pAG-MNase 酶进入细胞和胞核。因此,缓冲液中有足量的洋地黄皂苷对抗体和酶的成功结合以及靶向基因位点的消化至关重要。不同细胞系对毛地黄皂苷透化细胞显示不同的敏感性。虽然本实验步骤中建议的洋地黄皂苷量应足以对大多数细胞系或组织进行透化,但您可以使用本实验步骤测试您的特定细胞系或组织。我们发现,添加过量的洋地黄皂苷对本实验没有损害,因此无需生成浓度曲线。所以,进行快速测试以确定建议的洋地黄皂苷量是否适合你的细胞系就足够了。
注意:洋地黄皂苷溶液 #16359 应保存在 -20°C 下。请在使用过程中将其保存在冰上,并在实验完成后保存在 -20°C 下。
注意:如果肉眼看不到细胞沉淀,我们建议在步骤 2 中对细胞悬浮液进行初始离心后,在不干扰细胞沉淀的情况下去除尽可能多的细胞培养基,并对每个反应留下一些细胞培养基。然后在步骤 3 中,向细胞悬液中加入足够的 1X 洗涤缓冲液,使总体积达到 100 µl。
建议对input DNA 样品进行超声处理,因为 DNA 纯化离心柱只能纯化片段化的基因组 DNA (< 10 kb)。此外,片段化的基因组 DNA (< 1 kb) 可在 NG-seq 分析中用作阴性对照。应优化超声处理,以便input DNA 的长度为 100-600 bp。
我们建议使用input样品进行 NG-seq,因为它能方便且无偏倚的呈现细胞基因组。虽然 IgG 样品也可在 NG-seq 中用作阴性对照,但由于存在非特异性结合,它可能会显示基因组特定区域出现富集。非片段化input DNA 可用于 qPCR 分析。但必须使用苯酚/氯仿提取后再行乙醇沉淀方法来纯化非片段化的 DNA。
! 所有缓冲液的体积都应根据正在制备的input样品的数量按比例增加。
注意:如果在处理低细胞数(<100,000 个细胞)时肉眼看不到离心后的细胞沉淀,我们建议跳过下面的洗涤步骤 3-5。在步骤 2 中对细胞悬浮液进行初始离心后,在不干扰细胞沉淀的情况下去除尽可能多的细胞培养基,并在每个反应中留下一些细胞培养基。然后在步骤 6 中,将足够的 1X 洗涤缓冲液添加到细胞悬液中,以便在每个被测试的超声处理条件下达到 100 µl 的体积。
主要:在步骤 9 中,在 55°C 下孵育样品,因此建议使用一个可封盖的 1.5 ml 管子,以减少孵育过程中出现蒸发。
有关详细的疑难解答指南,请访问 https://cst-science.com/troubleshooting-CUT-RUN
实验步骤编号:1884
| ! | ! 表示实验步骤中需要根据进行的 CUT&Tag 反应次数来调整体积的重要步骤。 |
| !! | !! 表示需要在操作前稀释缓冲液的一个重要步骤。 |
| 安全停止 | 如果需要停止,这是实验步骤中的一个安全停止点。 |
! 所有缓冲液的体积都应根据正在进行的 CUT&Tag 反应次数按比例增加。
将 Concanavalin A Bead Activation Buffer 放在冰上。
确定需进行的 CUT&Tag 反应次数。我们强烈建议包含阳性对照 Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit mAb #9751 的反应。阴性对照 Normal Rabbit IgG #2729 或 Normal Mouse IgG #68860 是可选的,取决于要使用的峰值调用算法的要求。
通过轻轻上下吹打小心地把 Concanavalin A 磁珠重新悬浮到均匀的浆液中,确保不要将任何磁珠悬浮液溅出试管。
注:在整个实验步骤中避免将 Concanavalin A 磁珠悬浮液涡旋,因为反复涡旋可能会使 Concanavalin A 从珠子中移位。
按照每个 CUT&Tag 反应,将 10 µl 珠子悬浮液转移到一个新的 1.5 ml 微量离心管中。如果计划一次进行多次 14 CUT&Tag 反应,请使用两个或更多个 1.5 ml 微量离心管。每 1.5 ml 微量离心管中应加入不超过 140 μl 的 Concanavalin A 珠子。
每 10 µl 珠子添加 100 µl Concanavalin A Bead Activation Buffer。上下吸打来轻轻混合珠子。
将试管在磁力架上放置 30 秒钟至 2 分钟,然后使用移液管去除上清液。
注:为避免丢失珠子,在整个实验步骤中请勿使用真空来抽吸。
从磁力架上取下试管。重复步骤 4 和 5 来二次洗涤珠子。
添加一体积的 Concanavalin A Bead Activation Buffer,体积等同于初始的重悬珠子的液体体积(每次反应 10 µl),上下吹打重悬。
注:活化的珠子可在冰上储存长达 8 小时。
对于大多数细胞类型,活细胞可用于 CUT&Tag 检测,以产生强大的组蛋白、转录因子和辅因子富集。我们强烈建议尽可能使用活细胞。对于某些脆弱或对 Concanavalin A 敏感的细胞类型,请参阅附录 A,以了解 CUT&Tag 之前的细胞光固定实验步骤。请注意,细胞固定不会增加 CUT&Tag 信号,过度固定可能对酶切法片段化(Tagmentation)反应有害。
新鲜组织也可用于 CUT&Tag 测定,以产生大量的组蛋白富集。然而,转录因子和辅因子等非组蛋白靶标在 CUT&Tag 检测中并未得到很好的富集。为了分析组织中的转录因子和辅因子,我们建议使用 CUT&RUN Assay Kit #86652。新鲜组织通常会产生与固定组织相当或更强的 CUT&Tag 信号。如果需要固定,请参阅附录 B,以了解 CUT&Tag 实验之前组织的光固定实验步骤。
我们的 CUT&Tag 检测适用于各种不同的细胞和组织类型以及各种起始材料量。我们建议每次反应使用 100,000 个细胞或 1 mg 组织。如果起始细胞数量有限,则组蛋白修饰靶标每次反应可能只需要 5,000 到 10,000 个细胞,转录因子和辅因子每次反应可能需要 20,000 个细胞。低输入反应的成功取决于靶标丰度和抗体敏感性。足够量的起始材料对于所需的 CUT&Tag 信号至关重要,特别是对于转录因子和辅因子。每次反应最多可使用 250,000 个细胞或 5 mg 组织。一次反应中使用的整个实验步骤的缓冲液体积不需要根据细胞数量或组织质量进行调整,只要这些值落在指定范围内(5,000-250,000 细胞或 1-5 mg 组织)即可。当有说明时,所有缓冲液的体积需要根据正在进行的反应次数按比例调整。
推荐用于细胞透化的毛地黄皂苷的量过多,应该足以用于大多数细胞系和组织类型的透化。然而,并非所有细胞系和组织对毛地黄皂苷都表现出相同的敏感性。如果您的特定细胞系或组织在推荐的洋地黄皂苷浓度下不起作用,您可以按照附录 C 中提供的实验步骤来优化条件。洋地黄皂苷处理应导致 >90% 的细胞群透化。
! 所有缓冲液的体积都应根据正在进行的 CUT&Tag 反应次数按比例增加。
使用前,彻底解冻 200X Protease Inhibitor Cocktail #7012 和 100X Spermidine #27287,当天完成后,将其储存在 -20°C 下。请注意,由于含有 DMSO,Protease Inhibitor Cocktail #7012 置于冰上时会重新冻结。
制备完整的洗涤缓冲液(每个细胞制备物为 2 ml,每个 CUT&Tag 反应额外添加 100 μl),并在室温下保存。例如,如果同时使用未经处理和经药物处理的细胞(2 个细胞制备物)并进行 4 个抗体检测(阳性对照 H3K4me3 #9751、阴性对照 IgG #2729 或 #68860 以及两种实验抗体;8 次反应),则总共需要 4.8 ml 完整的洗涤缓冲液。
|
完全洗涤缓冲液 |
体积(每份细胞制备物) |
体积(每个反应) |
总体积 |
|
10X Wash buffer (CUT&RUN, CUT&Tag) #31415 |
200 µl |
10 μl |
将两列相加,得出所需的每种试剂总体积。 |
|
100X Spermidine #27287 |
20 µl |
1 µl |
|
|
Protease Inhibitor Cocktail (200X) #7012 |
10 μl |
0.5 μl |
|
|
Nuclease free water #12931 |
1770 μl |
88.5 μl |
注:活性细胞制备的所有步骤应在室温下连续进行,以尽量减少对细胞的压力。请勿涡旋细胞样品,以避免 DNA 片段化和细胞空化。
在室温下针对每次反应收集 100,000 个活性细胞,以尽量减少对细胞的压力。
注:对于贴壁细胞,使用胰蛋白酶将其从培养皿中分离,并用至少 3 个体积的组织培养基进行中和。我们不建议从培养皿中刮下细胞,以防止细胞裂解。应使用血细胞计数器或其他细胞计数器对细胞进行准确计数,以确保用于实验的细胞数量正确无误。
将细胞悬液在室温以 600 x g 离心 3 分钟,然后移除上清液。
注:如果使用的细胞数量少于 100,000 个总细胞,并且肉眼看不到离心的细胞沉淀物,我们建议跳过下面的洗涤步骤 3 至 5,直接进入步骤 6。在步骤 2 中对细胞悬浮液进行初始离心后,去除大部分上清液,每次反应留下 ≤40 µl 上清液。然后在步骤 6 中,向细胞悬浮液中添加足够的完整洗涤缓冲液,以使每次反应的总体积达到 100 µl。
轻轻上下摇晃,于室温下在 1 ml 完整洗涤缓冲液中重悬细胞沉淀物。
在室温下以 600 x g 的速度离心分离 3 分钟,然后移除上清液。
重复步骤 3 和 4 一次,以再次清洗细胞沉淀物。
针对每次反应,添加 100 µl 完整洗涤缓冲液,并轻轻上下摇晃移液管来重悬细胞沉淀物。
立即进入第 III 部分。
! 所有缓冲液的体积都应根据正在进行的 CUT&Tag 反应次数按比例增加。
使用前,彻底解冻 200X Protease Inhibitor Cocktail #7012 和 100X Spermidine #27287,当天完成后,将其储存在 -20°C 下。请注意,由于含有 DMSO,Protease Inhibitor Cocktail #7012 置于冰上时会重新冻结。
制备完整的洗涤缓冲液(每种组织类型制备 3 ml,每个反应额外添加 100 μl),并在室温下保存,以尽量减少对细胞的压力。例如,如果使用野生型和转基因肝脏作为起始材料(2 种组织类型)并进行 4 个抗体检测(阳性对照 H3K4me3 #9751、阴性对照 IgG #2729 或 #68860 以及两种实验抗体;8 次反应),则总共需要 6.8 ml 完整的洗涤缓冲液。
|
完全洗涤缓冲液 |
体积(每个组织类型) |
体积(每个反应) |
总体积 |
|
10X Wash buffer (CUT&RUN, CUT&Tag) #31415 |
300 µl |
10 μl |
将两列相加,得出所需的每种试剂总体积。 |
|
100X Spermidine #27287 |
30 µl |
1 µl |
|
|
Protease Inhibitor Cocktail (200X) #7012 |
15 μl |
0.5 μl |
|
|
Nuclease free water #12931 |
2655 μl |
88.5 μl |
针对每个反应称取 1 mg 新鲜组织。
将组织样本放入盘中,并使用干净的手术刀或剃须刀片切碎。将培养皿放在冰上。重要的是将组织置于低温下以防止蛋白质降解。
将组织重悬在 1 ml 完全洗涤缓冲液中,并将样品转移到 Dounce 匀浆器中。
用 20-25 个冲程将组织块分解成单细胞悬浮液,直到观察不到组织块。
将细胞悬液转移到 1.5 ml 管中,并在室温下以 3,000 x g 的速度离心 3 分钟,并用移液管从细胞中去除上清液。
在 1 ml 洗涤缓冲液中重悬细胞沉淀物。
将细胞悬液在室温以 3,000 x g 离心 3 分钟,然后移除上清液。
重复步骤 6 和 7 一次,以再次清洗细胞沉淀物。
针对每次反应,添加 100 µl 完整洗涤缓冲液,并轻轻上下摇晃移液管来重悬细胞沉淀物。
立即进入第 III 部分。
注:对于第 III-V 部分中的所有孵育步骤,无需通过摇动或旋转来混合样品。相反,我们建议只需将样品试管放在指定温度的架子上即可。在孵育步骤中混合样品不会提高测定的性能。相反,旋转或摇动样品,珠子可能会粘附在管壁和盖子上,从而导致更多的珠子结块和损失。
! 所有缓冲液的体积都应根据正在进行的 CUT&Tag 反应次数按比例增加。
在 90-100°C 下将 Digitonin Solution #16359 加热 5 分钟,并确保完全解冻并溶解。使用过程中,立即将解冻后的 Digitonin Solution #16359 置于冰上。当天完成后,储存在 -20°C 下。
使用前,彻底解冻 200X Protease Inhibitor Cocktail #7012 和 100X Spermidine #27287,当天完成后,将其储存在 -20°C 下。请注意,由于含有 DMSO,Protease Inhibitor Cocktail #7012 置于冰上时会重新冻结。
每次反应制备 100 μl 完整的抗体结合缓冲液并置于冰上。
|
完整的抗体结合缓冲液 |
体积(每个反应) |
|
Antibody Binding Buffer (CUT&RUN, CUT&Tag) #15338 |
96 μl |
|
100X Spermidine #27287 |
1 µl |
|
Protease Inhibitor Cocktail (200X) #7012 |
0.5 µl |
|
Digitonin Solution #16359 |
2.5 µl |
通过轻轻地上下吹打,彻底混合第 I 部分步骤 7 中准备的活化的 Concanavalin A 珠子。在 II-A 部分步骤 6 或 II-B 部分步骤 9 中制备的洗涤细胞悬浮液中,应添加珠子悬浮液。
将样品在室温下孵育 5 分钟。
将试管在磁力架上放置 30 秒钟至 2 分钟,然后取出并丢弃上清液。
从磁力架上取下试管。每次反应添加 100 μl 完整的抗体结合缓冲液,并通过移液管轻轻混合。
按每次反应分装 100 µl 细胞珠子悬液到一根单独的 1.5 ml 试管中,并放在冰上。
按每次反应添加适量的一抗,并上下吹打来轻柔混合。
注:CUT&Tag 所需的抗体量各不相同,应由用户确定。对于阳性对照 Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit mAb #9751 或阴性对照 Normal Rabbit IgG # 2729或 Normal Mouse IgG #68860,向反应中添加 2 μl 抗体。如果可能,我们强烈建议在检测中使用经 CUT&Tag 验证的抗体。CST 提供一系列经 CUT&Tag 验证的抗体,包括支持数据和适当的稀释比例。
在室温下孵育样品 1 小时。该步骤可以延长到在 4°C 下过夜。
立即进行第四部分。
! 所有缓冲液的体积都应根据正在进行的 CUT&Tag 反应次数按比例增加。
在 90-100°C 下将 Digitonin Solution #16359 加热 5 分钟,并确保完全解冻并溶解。使用过程中,立即将解冻后的 Digitonin Solution #16359 置于冰上。当天完成后,储存在 -20°C 下。
使用前,彻底解冻 200X Protease Inhibitor Cocktail #7012 和 100X Spermidine #27287,当天完成后,将其储存在 -20°C 下。请注意,由于含有 DMSO,Protease Inhibitor Cocktail #7012 置于冰上时会重新冻结。
每次反应新鲜制备 1.05 ml 洋地黄皂苷缓冲液,然后置于冰上。
NOTE: Digitonin Buffer prepared here will be used for both Section IV and V.
|
洋地黄皂苷缓冲液 |
体积(每个反应) |
|
10X Wash buffer (CUT&RUN, CUT&Tag) #31415 |
105 μl |
|
100X Spermidine #27287 |
10.5 μl |
|
Protease Inhibitor Cocktail (200X) #7012 |
5.25 μl |
|
Digitonin Solution #16359 |
26.25 μl |
|
Nuclease free water #12931 |
903 μl |
进行二抗预混合。对于每次反应,将 1 μl Goat Anti-Rabbit IgG (H+L) Antibody #35401 或 1 µl Donkey Anti-Mouse IgG (H+L) Antibody #52885 稀释至 50 μl 洋地黄皂苷缓冲液中。根据反应次数按比例增加二抗预混液。轻轻地上下吹打混合,然后置于冰上。
将含有第 III 部分步骤 7 一抗孵育溶液的样品试管在磁力架上放置 30 秒至 2 分钟,然后去除上清液。
向每个样品试管添加 50 µl 二抗预混合物,并轻轻上下吹打来混合样品。
室温下孵育样品 30 分钟。
立即进入第五部分。
! 所有缓冲液的体积都应根据正在进行的 CUT&Tag 反应次数按比例增加。
确保 10% SDS Solution #20533 完全溶于溶液中。在 37°C 下加热有助于溶解 SDS 沉淀物。
在 90-100°C 下将 Digitonin Solution #16359 加热 5 分钟,并确保完全解冻并溶解。使用过程中,立即将解冻后的 Digitonin Solution #16359 置于冰上。当天完成后,储存在 -20°C 下。
使用前,彻底解冻 200X Protease Inhibitor Cocktail #7012 和 100X Spermidine #27287,当天完成后,将其储存在 -20°C 下。请注意,由于含有 DMSO,Protease Inhibitor Cocktail #7012 置于冰上时会重新冻结。
每次反应制备 1.2 ml 高盐洋地黄皂苷缓冲液并置于冰上。
|
高盐洋地黄皂苷缓冲液 |
体积(每个反应) |
|
10X 高盐洗涤缓冲液(CUT&Tag) |
120 μl |
|
100X Spermidine #27287 |
12 µl |
|
Protease Inhibitor Cocktail (200X) #7012 |
6 µl |
|
Digitonin Solution #16359 |
30 µl |
|
Nuclease free water #12931 |
1032 µl |
每次反应制备 150 μl 酶切法片段化(Tagmentation)缓冲液并置于冰上。
|
酶切法片段化(Tagmentation)缓冲液 |
体积(每个反应) |
|
高盐洋地黄皂苷缓冲液(如上所述) |
148.5 µl |
|
氯化镁 |
1.5 µl |
对于每次反应,通过将 2 µl CUT&Tag pAG-Tn5 (Loaded) #79561 稀释至 50 μl 高盐洋地黄皂苷缓冲液中,来制备 pAG-Tn5 预混液。根据反应次数按比例增加 pAG-Tn5 预混液。轻轻地上下吹打混合,然后置于冰上。
将含有第 IV 部分步骤 4 一抗孵育溶液的样品试管在磁力架上放置 30 秒至 2 分钟,然后去除上清液。
从磁力架上取下试管,加入 500 µl 在第 IV 部分中制备的洋地黄皂苷缓冲液。轻轻上下吹打重悬珠子。
将试管在磁力架上放置 30 秒钟至 2 分钟,然后去除上清液。
重复步骤 3 和 4 一次,以进行第二次洗涤。
从磁力架上取下试管。向每根试管添加 50 µl pAG-Tn5 预混合物,并轻轻上下吹打来混合样品。
在室温下孵育 1 小时。
将试管在磁力分离架上放置 30 秒钟至 2 分钟,然后去除上清液。
从磁分离架上取下试管。添加 500 µl 高盐洋地黄皂苷缓冲液,并轻轻上下吹打来重悬珠子。
将试管在磁力架上放置 30 秒钟至 2 分钟,然后去除上清液。
重复步骤 9 和 10 一次,以进行第二次洗涤。
从磁力架上取下试管。向每支试管添加 150 μl 酶切法片段化(Tagmentation)缓冲液,并上下吹打以混合。
在 37°C 下孵育样品 1 小时。
要停止酶切法片段化(Tagmentation),向每个样品中添加 6.75 μl 0.5 M EDTA #7011、8.25 μl 10% SDS #20533和 1.5 μl 20 mg/mL Proteinase K #10012,然后快速涡旋以混合。
将样品在 58°C 下孵育 1 小时,将标记的染色质片段释放到溶液中。这次孵育可以延长过夜。如果孵育过夜,请使用有安全锁的试管以防止样品蒸发。
注:如果从固定细胞或组织开始,则在 65°C 下将样品置于有安全锁的试管中孵育 2 小时,以充分解交联。这种孵育可以延至过夜。
在室温下以 16,000x g 的速度离心 2 分钟,然后将试管置于磁力架上 30 秒钟至 2 分钟。
将上清液转移到新的 1.5 ml 试管中。这些是要纯化的 CUT&Tag DNA 样品。
继续执行第 VI 部分。(安全停止)或者,样品可以在 -20°C 下保存长达 1 周。然而,在 DNA 纯化之前,请务必将样品加热至室温(第 VI 部分)。
使用前请将 DNA 纯化柱、DNA 结合缓冲液、DNA 洗涤缓冲液和 DNA 洗脱缓冲液平衡至室温。
!!使用前,向 DNA 洗涤缓冲液中添加 24 ml 乙醇 (96-100%)。此步骤只需在第一组 DNA 纯化之前执行一次。
对于每个待纯化的 CUT&Tag DNA 样品,使用一个 DNA 纯化收集管。
向每份 CUT&Tag DNA 样品添加 833 μl DNA 结合缓冲液,通过上下吹打以混合。
注:每 1 体积的样品应使用 5 体积的 DNA 结合缓冲液。
从在步骤 1 中制备的每份样品中取出 600 μl 并将其至收集管的 DNA 离心柱中。
用离心机以 18,500 x g 离心 30 秒。
从收集管取出离心柱并丢弃液体。将离心柱放回空的收集管。
重复步骤 2-4,直到步骤 1 中的整个样品已经过离心柱旋转。将离心柱放回空的收集管。
向收集管的离心柱添加 750 μl DNA Wash Buffer。
用离心机以 18,500 x g 离心 30 秒。
从收集管取出离心柱并丢弃液体。将离心柱放回空的收集管。
用离心机以 18,500 x g 离心 30 秒。
丢弃收集管和液体。保留离心柱。
向每根离心柱添加 30 μl DNA 洗脱缓冲液,并放在干净的 1.5 ml 管中。
注:为了从柱中完全洗脱 DNA,需要最小体积为 30 µl 的 DNA 洗脱缓冲液。
用微量离心机以 18,500 x g 的离心力离心分离 30 秒,以洗脱 DNA。
取出并丢弃 DNA 旋转柱。洗脱液现在是纯化的 DNA。(安全停止)样品可在 -20°C 下保存长达 6 个月。
注:考虑到 CUT&Tag DNA 的产量通常较低,我们强烈建议使用所有 30 µl DNA 样品进行文库扩增。
使用该试剂盒制备的免疫富集的 DNA 样品与 NG-seq 直接兼容。对于下游 NG-seq DNA 文库构建,请使用与下游测序平台兼容的 DNA 文库制备实验步骤或试剂盒。对于 Illumina Systems 平台上的测序,我们建议使用 CUT&Tag Dual Index Primers and PCR Master Mix for Illumina #47415。请注意,DNA Library Prep Kit for Illumina Systems (ChIP-seq, CUT&RUN) #56795 和 Multiplex Oligos for Illumina Systems (ChIP-seq, CUT&RUN) #29580 或 #47538 与 CUT&Tag DNA 样品不兼容。
DNA 文库制备的其他建议:
已扩增的 CUT&Tag DNA 文库的产量可能因所使用的 DNA 定量方法而异。如果使用 Nanodrop 或 QIAxpert 系统,组蛋白靶标的预期读数为 10-20 ng/μl,非组蛋白靶标的预期读数为 5-12 ng/µl。如果 Nanodrop 或 QIAxpert 系统的文库浓度低于 3 ng/µl,请在对样品进行测序之前参阅疑难解答指南。如果使用 Qubit 荧光定量系统或 Picogreen 检测法,组蛋白靶标的预期读数为 3-10 ng/μl,非组蛋白靶标的预期读数可能低于 1 ng/µl。由于浓度较低,Bioanalyzer 或 TapeStation 系统生成的谱图中可能会看到极弱的峰甚或看不到可见峰,特别是对于细胞中不丰富的靶标。在这些情况下,如果阳性对照 Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit mAb #9751 产生预期的文库产量和/或 Bioanalyzer 峰,这表明在总体上来讲实验是成功的,我们仍然建议继续进行 NGS。请参阅我们的 CUT&Tag 常见问题解答网页以获取支持数据,或需要额外的指导来汇集各种产量的样品。
无论靶标类型如何,每个样本 200 万个读数的测序深度通常足以进行 CUT&Tag 测定。如果每份样品的测序深度大于 1500 万,则读取的重复率显著增加。如果每份样品的测序深度低于一百万个读数,则信噪比会降低。
对于少于 20,000 个起始细胞数,在 NGS 读取中获得较低的映射率或较高的重复率是很常见的。如果发生这种情况,我们建议增加测序深度以获得足够的唯一映射读取用于下游数据分析。
我们强烈建议尽可能使用活细胞。对于某些对伴刀豆球蛋白 A 脆弱或敏感的细胞类型,请在 CUT&Tag 实验之前参考下面的实验步骤略微固定细胞。请注意,细胞固定并不显著增加 CUT&Tag 信号。事实上,过度固定可能导致 CUT&Tag 信号更弱。请参阅第 II 部分中的描述,以确定每个反应中相应的细胞数量。
注:固定细胞制备需要以下试剂,不包含在此试剂盒中:37% 甲醛或 16% 无甲醇的甲醛 #12606,甘氨酸溶液 (10X) #7005。
! 所有缓冲液的体积都应根据正在进行的 CUT&Tag 反应次数按比例增加。
每 1 ml 待处理细胞悬液,配制 2.7 µl 37% 甲醛液或 6.25 µl 16% Formaldehyde, Methanol-Free #12606 并室温保存。使用制造商规定的有效期内的新鲜甲醛。
使用前,彻底解冻 200X Protease Inhibitor Cocktail #7012 和 100X Spermidine #27287,当天完成后,将其储存在 -20°C 下。请注意,由于含有 DMSO,Protease Inhibitor Cocktail #7012 置于冰上时会重新冻结。
制备完整的洗涤缓冲液(每个细胞制备物为 2 ml,每个 CUT&Tag 反应额外添加 100 µl)并将其保存在室温下。例如,如果同时使用未经处理和经过药物处理的细胞(2 个细胞制备物)并进行 4 个抗体检测(阳性对照 H3K4me3 #9751、阴性对照 IgG #2729 或 #68860,以及两种实验抗体;8 次反应),则总共需要 4.8 ml 完整洗涤缓冲液。
|
完全洗涤缓冲液 |
体积(每份细胞制备物) |
体积(每个反应) |
总体积 |
|
10X Wash buffer (CUT&RUN, CUT&Tag) #31415 |
200 µl |
10 μl |
将两列相加,得出所需的每种试剂总体积。 |
|
100X Spermidine #27287 |
20 µl |
1 µl |
|
|
Protease Inhibitor Cocktail (200X) #7012 |
10 μl |
0.5 μl |
|
|
Nuclease free water #12931 |
1770 μl |
88.5 μl |
每个反应收获 100,000 个活细胞。
注:对于贴壁细胞,使用胰蛋白酶将其从培养皿中分离出来,并用至少 3 个体积的组织培养基进行中和。我们不建议从培养皿中刮下细胞,以防止细胞裂解。应使用血细胞计数器或其他细胞计数器对细胞进行准确计数,以确保用于实验的细胞数量正确无误。
每 1 ml 细胞悬液添加 2.7 µl 37% 甲醛或 6.25 µl 16% 无甲醇的甲醛 #12606,以实现 0.1% 甲醛终浓度。旋转混匀并置于室温下孵育 2 分钟。
每 1 ml 固定细胞悬浮液中添加 100 µl Glycine Solution (10X) #7005 来停止交联。旋转试管以混合并在室温下孵育 5 分钟。
在 4°C下以 3,000 x g 的离心速度把细胞悬浮液离心 3 分钟,并除去上清液。立即进行步骤 5。(安全停止)或者,固定的细胞沉淀物在使用前可在 -80°C 下保存长达 6 个月。
注:如果使用的细胞少于 100,000 个总细胞,并且肉眼看不到离心的细胞沉淀物,我们不建议冷冻细胞沉淀物。相反,我们建议继续执行该实验步骤并跳过下面的洗涤步骤 5 至 7。在步骤 4 中对细胞悬浮液进行初始离心后,去除大部分上清液,每次反应留下 ≤40 µl 细胞培养基。然后在步骤 8 中,向细胞悬浮液中添加足够的完整洗涤缓冲液,以使每次反应的总体积达到 100 µl。
通过柔和上下吹打,在 1 ml 完全洗涤缓冲液中重悬细胞沉淀物。
在 4°C 下以 3,000 x g 的速度离心 3 分钟,然后移除上清液。
重复步骤 5 和 6 一次,以再次清洗细胞沉淀物。
针对每个反应,添加 100 µl 完全洗涤缓冲液,并通过柔和上下吹打来重悬细胞沉淀物。
立即进入第 III 部分。
对于大多数组织类型,1 mg 的新鲜组织足以产生组蛋白的强烈富集。如果无法获得新鲜组织,可以使用轻微固定的组织(0.1% 甲醛需 2 分钟)。固定的组织样品在使用前可在 -80°C 下冷冻长达 6 个月。过度固定可能会导致 CUT&Tag 信号变弱。CUT&Tag 测定不能很好地富集组织中的转录因子和辅因子。对于转录因子和辅因子的分析,我们建议使用 CUT&RUN Assay Kit #86652。
注:固定组织制备需要以下试剂,不包含在此试剂盒中:37% 甲醛或 16% 无甲醇的甲醛 #12606、磷酸盐缓冲盐水 (PBS) #9872 和甘氨酸溶液 (10X) #7005。
! 所有缓冲液的体积都应根据正在进行的 CUT&Tag 反应次数按比例增加。
每 1 ml 待处理细胞悬液,配制 2.7 µl 37% 甲醛液或 6.25 µl 16% Formaldehyde, Methanol-Free #12606 并室温保存。使用在制造商标示的有效期内的新鲜甲醛。
每 1 ml 固定缓冲液制备 100 µl 甘氨酸溶液 (10X) #7005。
使用前,彻底解冻 200X Protease Inhibitor Cocktail #7012 和 100X Spermidine #27287,当天完成后,将其储存在 -20°C 下。请注意,由于含有 DMSO,Protease Inhibitor Cocktail #7012 置于冰上时会重新冻结。
配制完全洗涤缓冲液(3 ml 用于每个组织类型,额外 100µl 用于每个反应 )并保存于室温,以最大限度减少细胞应激。
|
完全洗涤缓冲液 |
体积(每个组织类型) |
体积(每个反应) |
总体积 |
|
10X Wash buffer (CUT&RUN, CUT&Tag) #31415 |
300 µl |
10 μl |
将两列相加,得出所需的每种试剂总体积。 |
|
100X Spermidine #27287 |
30 µl |
1 µl |
|
|
Protease Inhibitor Cocktail (200X) #7012 |
15 μl |
0.5 μl |
|
|
Nuclease free water #12931 |
2655 μl |
88.5 μl |
针对每个组织类型配制 1 ml 固定缓冲液。使用制造商规定的有效期内的新鲜甲醛。
|
固定缓冲液 |
体积(每个组织类型) |
|
甲醛 |
2.7 µl 37% 或 6.25 µl 16% |
|
Protease Inhibitor Cocktail (200X) #7012 |
5 μl |
|
磷酸盐缓冲盐水 (PBS) #9872 |
992.3 μl |
针对每个组织类型配制 1 ml 固定洗涤缓冲液并置于冰上。
针对每个反应称取 1 mg 新鲜组织。
将组织样本放入盘中,并使用干净的手术刀或剃须刀片切碎。将培养皿放在冰上。重要的是将组织置于低温下以防止蛋白质降解。
将切碎的组织立即转移到 1 ml 固定缓冲液中,并旋转试管以混合。
注:此固定溶液体积足以容纳高达 50 mg 的组织。如果处理 >50 mg,则相应地增加步骤 3 中使用的固定缓冲液和步骤 7 中使用的固定洗涤缓冲液的量。
室温孵育 2 分钟。
每 1 ml 固定缓冲液中添加 100 µl 甘氨酸溶液 (10X) #7005 来停止交联。旋转试管以混合并在室温下孵育 5 分钟。
将组织在 4°C下以 2,000 x g 离心 5 分钟,然后移除上清液。
用 1 ml 固定洗涤缓冲液重悬组织。
在 4°C 下以 2,000 x g 的速度离心 5 分钟,然后去除上清液并继续执行步骤 9。(安全停止)或者,固定的组织沉淀物在分解前在 -80°C 下可储存长达 6 个月。
将组织重悬在 1 ml 完全洗涤缓冲液中,并将样品转移到 Dounce 匀浆器中。
用 20-25 冲程将组织块分解成单细胞悬浮液,直到观察不到组织块。
将细胞悬液转移到 1.5 ml 管中,并在室温下以 3,000 x g 的速度离心 3 分钟,从细胞中去除上清液。
在 1 ml 完全洗涤缓冲液中重悬细胞沉淀物。
将细胞悬液在室温以 3,000 x g 离心 3 分钟,然后移除上清液。
重复步骤 12 和 13 一次,以再次清洗细胞沉淀物。
针对每个反应,添加 100 µl 完全洗涤缓冲液,并通过柔和上下吹打来重悬细胞沉淀物。
立即进入第 III 部分。
在 CUT&Tag 实验步骤中,向缓冲液添加毛地黄皂苷促进了细胞膜透化以及一抗、二抗和 pAG-Tn5 酶进入细胞和胞核。因此,缓冲液中有足量的毛地黄皂苷对抗体与酶成功结合及消化靶向的基因座至关重要。不同细胞系对毛地黄皂苷透化细胞显示不同的敏感性。虽然本实验步骤中建议的洋地黄皂苷量应足以对大多数细胞系或组织进行透化,但您可以使用本实验步骤测试您的特定细胞系或组织。我们发现,添加过量的洋地黄皂苷对本实验没有损害,因此无需生成浓度曲线。所以,进行快速测试以确定建议的洋地黄皂苷量是否适合你的细胞系就足够了。
在 90-100°C 下将 Digitonin Solution #16359 加热 5 分钟,并确保完全解冻并溶解。使用过程中,立即将解冻后的 Digitonin Solution #16359 置于冰上。当天完成后,储存在 -20°C 下。
配制 100 μl 1X 洗涤缓冲液/反应。无需为这个测试在缓冲液中添加亚精胺或蛋白酶抑制剂。
在 1.5 ml 试管中,采集 100,000 个细胞(来自第 II-A 部分第 1 步),在室温下以 600 x g 的速度离心 3 分钟,并抽取上清液。对于组织,从 1 mg 组织中收集分解的细胞(第 II-B 部分步骤 1-8)。
在 100 µl 1X 洗涤缓冲液中重悬细胞沉淀物。
将 2.5 µl 洋地黄皂苷溶液 #16359 添加到每次反应中,并在室温下孵育 10 分钟。
将 10 µl 细胞悬液与 10 µl 0.4% 台盼蓝染料混合。
使用血细胞计数器或细胞计数器计算染色细胞的数量和细胞总数。充分通透会导致 > 90% 的细胞被台盼蓝染色。
如果被台盼蓝染色的细胞不到 90%,则增加每个反应中添加的洋地黄皂苷溶液 #16359 的量,并重复步骤 1-5,直到 > 90% 的细胞被透化和染色。在第 I - V 部分中采用上述洋地黄皂苷溶液 #16359 的量。
CUT&Tag 依照来自 Active Motif, Inc.的许可证、依照美国专利第 10,689,643 号和第 9,938,524 号、其外国等效专利及从中衍生的子专利提供。仅供购买者内部研究使用。不可用于转售、服务或其他商业用途。
美国专利第 11,733,248 号、外国等效专利及从中衍生的子专利。
实验步骤编号:2745
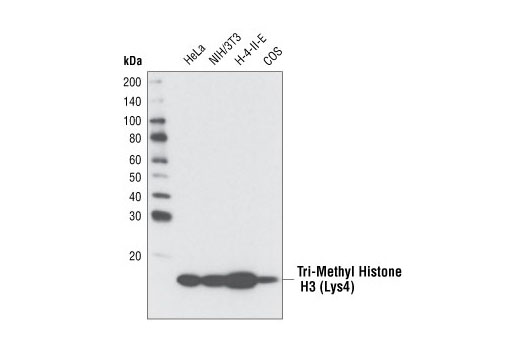
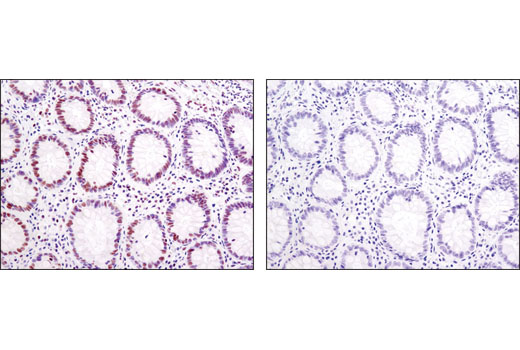
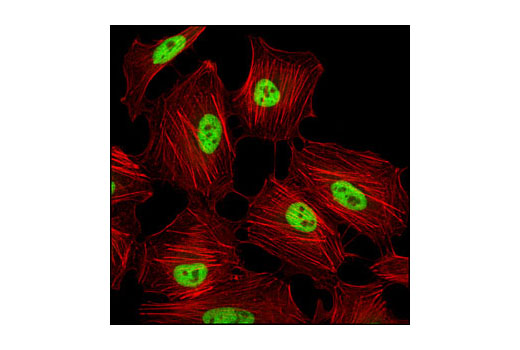
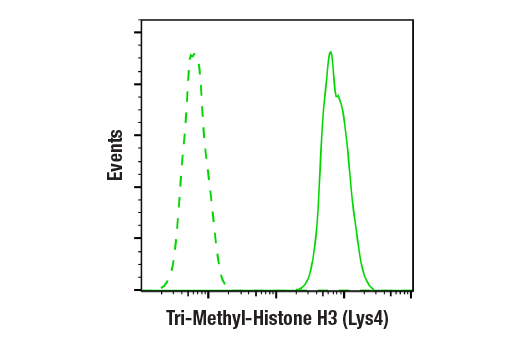
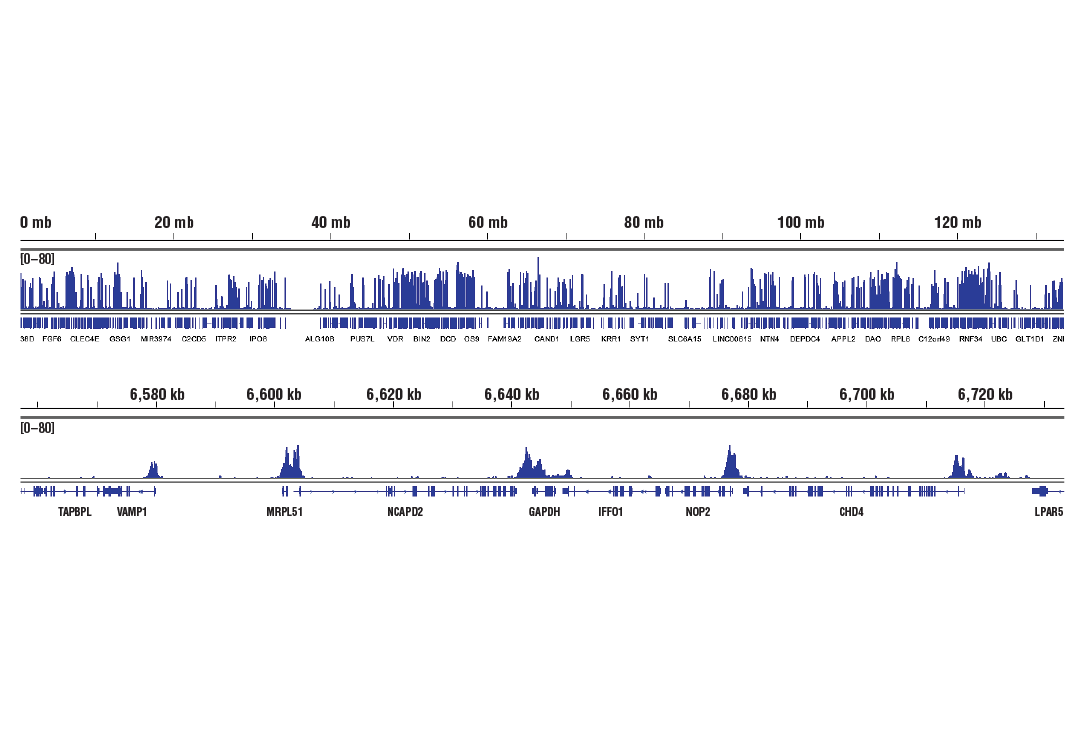
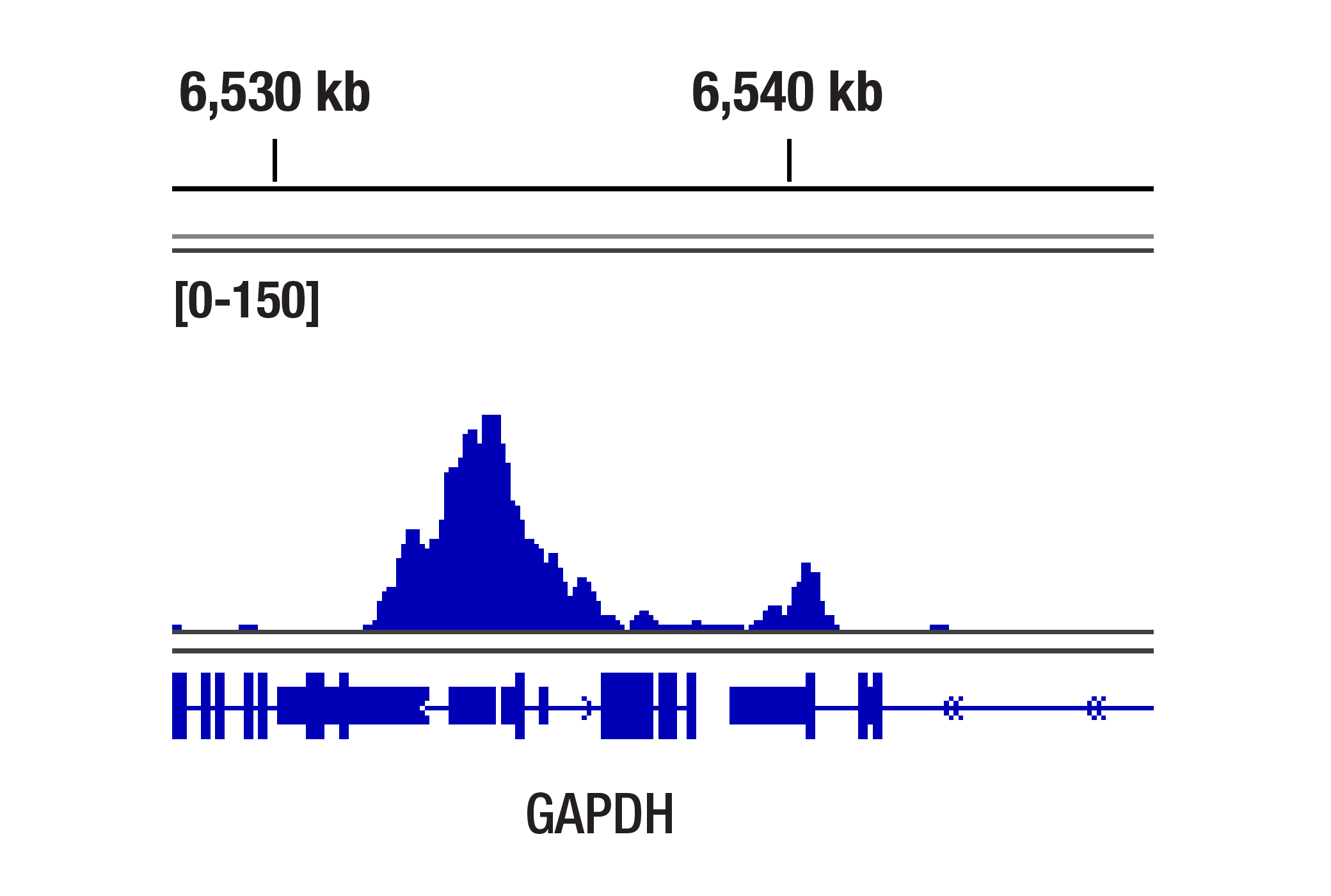
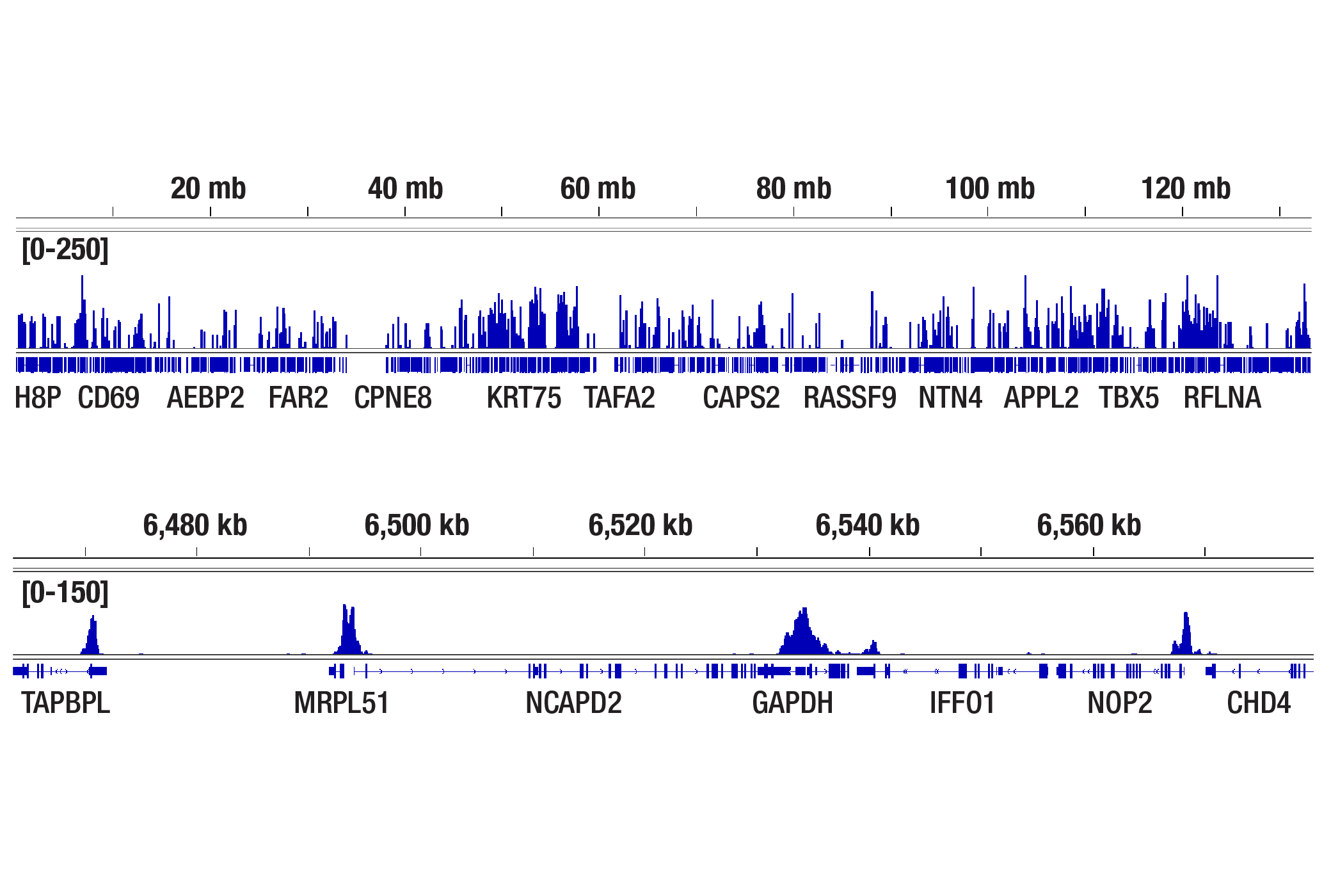
人, 小鼠, 大鼠, 猴, 黑腹果蝇 , 酿酒酵母
非洲爪蟾蜍, 斑马鱼
使用与 Lys4 三甲基化组蛋白 H3 氨基末端相对应的合成肽对动物进行免疫接种来产生单克隆抗体。
核小体由四个核心组蛋白(H2A、H2B、H3 和 H4)组成,是染色质的主要组成部分。最初被认为可作为 DNA 包装的静态支架的组蛋白现在已被证明是动态蛋白,可进行多种类型的翻译后修饰,包括乙酰化、磷酸化、甲基化和泛素化 (1)。组蛋白甲基化是形成基因组的活跃和非活跃区域的主要决定因素,并且在发育期间基因组的正确编程过程中发挥关键作用 (2,3)。组蛋白 H3 (Arg2, 17, 26) 和 H4 (Arg3) 的精氨酸甲基化可促进转录激活,并由蛋白精氨酸甲基转移酶 (PRMTs) 家族介导,该家族包括辅助激活因子 PRMT1 和 CARM1 (PRMT4) (4)。与此相反,已确定更多样化的组蛋白赖氨酸甲基转移酶,除了其中某个外其余的都含有一个保守催化 SET 结构域,该结构域最初在果蝇 Su(var)3-9、zeste 增强子和 Trithorax 蛋白中得以确认。赖氨酸甲基化主要发生在组蛋白 H3 (Lys4 位点, 9, 27, 36, 79) 和 H4 (Lys20 位点) 中,并已确定与转录激活和沉默有关 (4)。这些赖氨酸残基的甲基化可协调染色质修饰酶的募集,这些酶含有甲基赖氨酸结合模块,例如克罗莫结构域 (HP1, PRC1)、PHD fingers (BPTF, ING2)、tudor 结构域 (53BP1) 和 WD-40 结构域 (WDR5) (5-8)。PADI4、LSD1、JMJD1、JMJD2 和 JHDM1 等组蛋白脱甲基酶的发现表明:甲基化是可逆的表观遗传标记物 (9)。
探索与本品相关的通路。
STRING - 已知和预测的蛋白质间相互作用。
除非如以 CST 合法授权代表签署的书面形式另行明确同意,否则以下条款适用于 CST、其附属公司或其分销商提供的产品。除非 CST 合法授权代表以书面形式单独接受,否则任何附加于或异于此处所载条款和条件的客户条款和条件均被拒绝且无效。
产品用“仅供研究使用”或类似标示声明标示,并且尚未经 FDA 或其他国外或国内监管实体出于任何目的批准、准许或许可。客户不得出于任何诊断或治疗目的或以任何与产品标示声明相冲突的方式使用任何产品。CST 销售或许可的产品提供给作为最终用户的客户,且仅用于研究和开发用途。出于诊断、预防或治疗目的任何产品使用或出于转售(单独或作为成分)或其他商业目的的任何产品购买都要求来自 CST 的单独许可。客户 (a) 不得向任何第三方出售、许可、出借、捐赠或另行转让或提供任何本公司产品,无论单独或联合其他材料方式,或使用本公司产品制造任何商业产品,(b) 不得复制、修改、逆向工程、反编译、反汇编或另行尝试发现本公司产品的底层结构或技术,或出于开发与 CST 产品或服务竞争的任何产品或服务的目的使用本公司产品,(c) 不得从本公司产品改变或移除任何商标、商品名称、徽标、专利或版权声明或标记,(d) 仅应根据 CST 产品销售条款和任何适用文档使用本公司产品,以及 (e) 应就客户联系本公司产品所用的任何第三方产品或服务而言遵守任何许可、服务条款或类似协议。